
Introduction
In relation to population aging occurring globally, it is estimated that the number of people aged ≥ 60 years could double by 2050 (1). Given the numerous chronic diseases associated with advancing age, the risks of adverse drug reactions due to polypharmacy are increasing (2). In some cases, the extend of undesired side effects could be decreased by applying lower doses of active pharmaceutical ingredient (API), thereby also reducing the effect of pharmaceuticals on the environment, which is in line with the current trend toward green pharmacy (3). While for some APIs it is possible to reduce the dose directly without compromising therapeutic efficacy, other options would utilize different aspects of pharmacokinetics – in particular improving absorption of the API or retarding metabolism or excretion (4). As the proportion of drugs with poor aqueous solubility continuous to increase (5–7), different strategies for improving their solubility are of vital importance in drug development, especially for APIs with doses ≥ 50 mg, which are usually incompletely absorbed when administered orally. Following the commercial success of Sandimune Neoral® as a self-microemulsifying drug delivery system (SMEDDS) formulated with cyclosporine A, lipid-based systems (LBSs) have attracted considerable attention as a formulation approach for improving the bioavailability of API with poor biopharmaceutical properties (8).
This type of formulation has been on the market for more than 30 years, so it is no longer considered new. Nevertheless, the number of publications on LBSs is constantly increasing, together with the interest in developing them for clinical applications. The current state of patents for self(micro)emulsifying drug delivery systems was critically evaluated in the recent review (9). Over the last decade, research has focused on various (industrially applicable) solidification techniques that can be utilized to transform liquid SMEDDS into solids that merge the advantages of liquid LBSs and solid dosage forms (10–12) and are able to maintain API in a dissolved state not only in the formulation but also during passage through the gastrointestinal tract (13). The development of (solid) SMEDDS and their characterization techniques, which are beyond the scope of this review, have recently been described in detail by Rajpoot et al. (14), while current and future industrially applicable approaches to maintain the API in a solubilized state were summarized by Holm et al. (15). To maintain API in a dissolved state throughout gastrointestinal transit, so called supersaturable SMEDDS were introduced (16, 17) containing polymers capable of reducing the precipitation of API in vivo by maintaining the transient supersaturation of the drug that occurs after dispersion of LBSs in the gastrointestinal fluid . Holm et al. systematically presented also the design and evaluation of supersaturated LBSs, containing API in concentrations above thermodynamic solubility already in the formulation, together with the impact of both types of supersaturating LBSs on improved bioavailability of drugs formulated either as liquid SMEDDS or SMEDDS powders obtained by direct adsorption technique.
In the present review, the influence of different precipitation inhibitors on the precipitation of API is presented from a mechanistic point of view, which is particularly relevant for pharmaceutical technologists focusing on preformulation studies of supersaturable LBSs. In addition to self-emulsifying powders, self-emulsifying granules, and pellets with polymeric precipitation inhibitors were also discussed. Besides being patient-friendly dosage forms, multiparticulates can also be further processed into self-emulsifying tablets or hard capsules, which is in line with the ability of supersaturating LBSs to load the entire dose of API into the defined volumes of hard capsules (or a tablet of acceptable size).
LIPID-BASED SYSTEMS AS FORMULATION APPROACH TO IMPROVE THE API BIOAVAILABILITY AND DOSE LOWERING
By solubilizing the drug within SMEDDS, bioavailability could be improved not only by enhancing API aqueous solubility or ideally avoiding the dissolution step, but also by improving intestine permeability and inhibiting efflux proteins in addition to decreasing pre-systemic metabolism (6, 18, 19), as previously presented in the present journal (20). LBSs are therefore recognized as one of the most plausible platforms for the delivery of lipophilic but also hydrophobic APIs, and particularly emulsion-based systems are suitable for such purposes (21–23). In addition to facilitating the development of formulations for enhanced oral application and patient compliance, the use of SMEDDS for the delivery of therapeutic peptides and genes is also being investigated. Based on the recent research (24, 25).
S(M)EDDS demonstrates the capability to shield macromolecules from the harsh biological environment, thus the potential applications of this system are not limited to small molecule APIs.
According to the lipid-based classification system, LBSs are divided into 4 classes, with SMEDDS belonging to type IIIb systems (20, 26). It is defined as an isotropic mixture of oils, (hydrophilic) surfactants, co-surfactants, and hydrophilic cosolvents that spontaneously form oil-in-water (micro)emulsion with droplet size below 50 or 100 nm in the presence of aqueous media. In addition to self-emulsifying drug delivery systems (SEDDS) and self-nanoemulsifying drug delivery systems (SNEDDS), also known for their self-emulsifying ability, it is acknowledged as one of the most efficient in improving aqueous solubility and bioavailability of poorly water-soluble APIs (27). They have fairly similar compositions, varying in oil/surfactant ratio and lipophilicity. In the literature, the use of terminology with regard to SMEDDS and SNEDDS is rather inconsistent (28), therefore in the present review article, the naming SMEDDS will be used for all systems forming (micro)emulsion with droplet size below 100 nm on contact with aqueous milieu that can be classified as Type IIIb systems. On the other side, SEDDS could be classified either as Type II or IIIa system, depending on the hydro-lipophilicity of incorporated surfactants and proportion of lipids, with Type II formulation being less hydrophilic. Nevertheless, both SEDDS types form emulsions with lipid droplet size 100–300 (250) nm upon oral administration. Beside self-emulsifying formulations, type IV systems, as representatives of most hydrophilic LBSs composed only of surfactants and hydrophilic co-solvents, also self-disperse spontaneously in contact with aqueous media. However, in this case, dispersion of colloidal-size micelles is formed instead of (micro)emulsion. The risk of drug precipitation upon oral administration of LBSs increases with lower oil phase content and higher hydrophilicity of surfactants and/or cosolvents. In keeping with this, most hydrophilic formulations, i.e. Type IV systems closely followed by Type IIIB, are associated with a higher risk of incomplete drug absorption (26, 29–31).
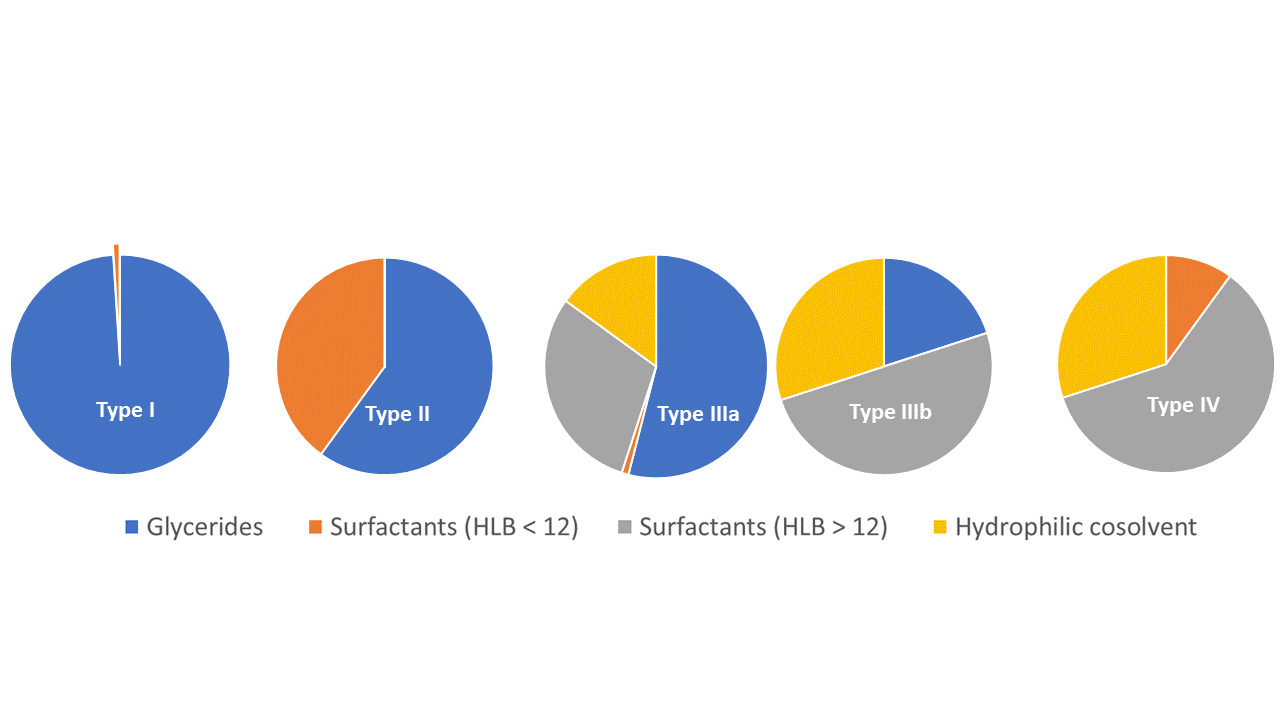
Fig. 1. Classification and approximate composition of lipid formulation system, based upon (26).
Upon oral administration, SMEDDS exhibit rapid self-emulsification in the aqueous media supported by agitation provided by natural motility of the gastrointestinal tract. Ideally, this process enables the API to remain dissolved within the resulting oil droplets of microemulsion (19). In reality, dilution of LBSs with gastrointestinal fluids is the first critical step possibly leading to API precipitation, in addition to the lipid digestion process, as API saturation and subsequent supersaturation are likely to occur (6).
Chemical digestion of lipids starts in the stomach by gastric lipases digesting the short- and medium-chain fatty acids, and proceeds in the duodenum under the action of pancreatic (co)lipases digesting also the long-chain triglycerides (32). In the intestine, API is then solubilized in a complex mixture of colloidal species, composed of (digested) components of SMEDDS, endogenous bile salts, and phospholipids as well as cholesterol. The risk for API precipitation from formed microemulsion depends on the physicochemical characteristic of API and lipids ( i.e. the chain-length of fatty acids esterified in the triglycerides effects) in addition to the increased polarity of products obtained through the lipid digestion process that negatively influences the ability of SMEDDS to incorporate the lipophilic drug (33). The solubility effect of LBS digestion products was studied by Alskär et al. (34) where weak bases were reported to show significantly increased solubility in the less lipophilic milieu upon lipid digestion as compared to the solubility of neutral and acidic API that decreases. In addition, the API solubilization capacity can also be reduced due to faster absorption of lipid-digestion products as compared to API, potentially resulting in exceeding saturation concentration (35). As high drug concentrations are the driving force not only for API precipitation but also for its flux across the GIT membrane, enhanced absorption could be reached after a sufficient supersaturation period. In keeping with this ideal, SMEDDS formulation should maintain a supersaturated state long enough to allow transport of the complete dose through the intestinal wall, which could be achieved with the support of polymeric precipitation inhibitors.
The importance of supersaturation induction and its maintenance through longer periods of time
The supersaturation state is favorable in terms of enhanced absorption and as such presents a good strategy for supersaturation that could be described with the supersaturation ratio (SR) expressed as the quotient of measured (supersaturated) API concentration at a determined time point ( ct) and equilibrium API solubility overcoming the challenge of low oral bioavailability of API with limited aqueous solubility ( i.e. BCS II and BCS IV drugs) as well as those with a high single dose. According to definition the supersaturation (36) occurs when API is present in the amount higher than its saturation solubility. The state of ( ceq) ( Equation 1):
SR = ( 1)
On this basis, saturated solutions are described with a value of SR = 1 and supersaturated with SR > 1 (37). Nevertheless, the supersaturated state tends to return to the equilibrium state by precipitation due to its thermodynamic instability. Since the molecules of the API move randomly within the homogenous supersaturated solution, with limited interparticulate space, they spontaneously associate in small clusters. In addition to active deposition on the neighboring surfaces, the nucleation process could also be triggered by the presence of various impurities, onto which the solute aggregates (38, 39), although the exact process of nucleation is still unclear (40). Regardless of the origin of nucleation, the formed cluster grows by further deposition of molecules on its surface until it reaches a critical size, which can then grow into ordered macroscopic crystals by becoming the center of crystallization. Three main theoretical models (the surface energy theory, the diffusion theory, and the adsorption layer theory) were introduced to describe the crystal growth process (39, 41–43). To avoid the crystallization process and take advantage of the induced supersaturated state, the process of API precipitation must be inhibited or delayed. The concept of generating and maintaining a metastable supersaturated state is commonly referred to as the “spring and parachute” theory (44–46). The state of supersaturation is usually generated by increasing the drug concentration beyond the saturation solubility, as described by the term “spring”. The degree of supersaturation is the initial driving force for precipitation – the higher the initial supersaturation, the faster precipitation occurs (47). By applying a precipitation inhibitor ( i.e. the “parachute”) supersaturation is maintained by stabilizing the metastable state produced. By slowly lowering the concentration to the saturation limit, the “parachute” excipient provides sufficient time for absorption, similar to how the parachute device slows the movement of an object through the atmosphere (48). The “spring” effect of API-loaded SMEDDS generating a metastable supersaturated state and the “parachute” effect supporting the prolonged API supersaturation achieved by the incorporation of (polymeric) precipitation inhibitors into such a formulation is schematically illustrated in Figure 2. For comparison, the drug dissolution profile of a conventional solid dosage form is also illustrated to depict the impact of solubilization and supersaturation effect achieved by simple API-loaded SMEDDS and SMEDDS with precipitation inhibitors.
In several studies conducted by Williams et al., the composition of different LBS formulations was tested in relation to the supersaturation effect. It was found that medium-chain LBSs were able to produce a higher degree of supersaturation compared to long-chain triglycerides, due to a higher API loading related to the better solubility of the hydrophobic API studied in medium-chain triglycerides and more water-soluble lipid-digestion products with lower solubilization capacity. This proved to be advantageous for highly permeable APIs, such as fenofibrate and tolfenamic acid, both being widely used in the elderly population for the treatment of dyslipidemia and pain-relief (49). However, the in vivo behavior of medium-chain and long-chain LBS is drug-specific and therefore cannot be considered as a rule.
Another study by Williams et al. investigated the API-, formulation- and dose-dependence in relation to the maintenance of supersaturation and precipitation potential during in vitro digestion testing. The authors were able to predict the probability of precipitation in vitro by observing the maximum supersaturation ratio SRM (maximum degree of supersaturation). The study revealed a threshold value of SRM equal to 3 (SRM = 3), as the tested LBSs with a value below 3 maintained supersaturation without signs of precipitation, while a higher supersaturation concentration (SRM> 3) led to precipitation of API. Therefore, the “favorable” supersaturation concentrations (SRM< 3) are more likely to enhance absorption in vivo, while formulations with SRM value above 3 may promote precipitation of API, resulting in incomplete bioavailability (50, 51).
In line with the “spring and parachute” strategy, drug development is focused on formulations that are capable not only of inducing the supersaturation state but also delaying and/or inhibiting the precipitation of API, thus supporting the beneficial effects of the supersaturation state. By designing such supersaturable delivery systems, it is possible to maintain this state for a limited time period, long enough to allow adequate absorption. In addition, such formulations allow a higher dose of API to be incorporated and absorbed with less risk of precipitation (16, 33, 36). Thus, the main advantage of supersaturable SMEDDS over the “ simple” SMEDDS is that they are able to deliver a larger amount of the in an equal or even smaller volume of SMEDDS, thus also delivering lower concertation of surfactant, which contributes to less irritation of the gastrointestinal tract.
As far as terminology is concerned, supersaturable formulations should not be misunderstood for supersaturated once . While supersaturated SMEDDS are formulations in which the API is incorporated in a concentration above the solubility limit, the API concentration in supersaturable SMEDDS becomes supersaturated upon dilution/dispersion of the formulation. Therefore, the API in supersaturated formulations may already precipitate during storage due to thermodynamic instability, while this is not the case in supersaturable systems (13).
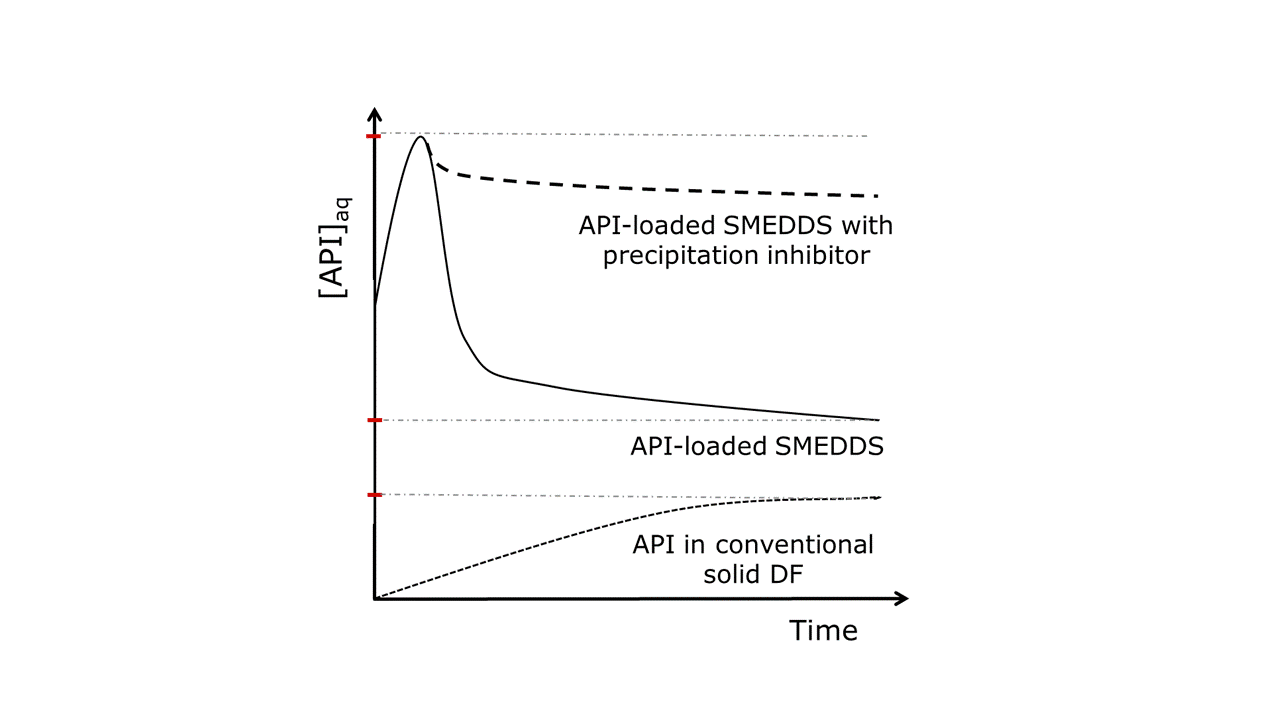
Fig. 2. The influence of the drug delivery systems (SMEDDS with or without precipitation inhibitors vs. conventional solid dosage form (DF)) on the dissolution profile and solubility of API in the gastrointestinal fluids is schematically presented, and the importance of precipitation inhibitors in supersaturating drug delivery systems is illustrated. Adapted from (18).
PRECIPITATION INHIBITION AND EXCIPIENTS USED TO STABILIZE SUPERSATURATION
Various excipients have been investigated as precipitation inhibitors in supersaturable formulations, of which polymeric molecules have been found to be the most efficient (38, 44, 52). They can inhibit either nucleation or crystal growth, both important steps in the crystallization process of API. It has been reported that polymers that can be dissolved in LBSs can function just as well as polymeric precipitation inhibitors when compared to those that are readily soluble in water (53, 54). The group of polymers most widely used to stabilize the supersaturation formed upon administration of SMEDDS include: 1) cellulose derivatives, e.g. hypromellose (HPMC), hydroxypropyl cellulose (HPC), hydroxyethyl cellulose (HEC); 2) vinyl polymers, e.g. polyvinypyrrolidone (PVP), polyvinyl alcohol (PVA), polyvinypyrrolidone/vinyl acetate copolymer (PVP/VA); 3) polyethylene glycol (PEG) copolymers; 4) polyoxyethylene copolymers; 5) polymetacrylates and 6) others (Fig. 3). Added in low concentrations, they significantly affect drug nucleation and/or crystal growth rates, leading to drug precipitation inhibition (37, 55).
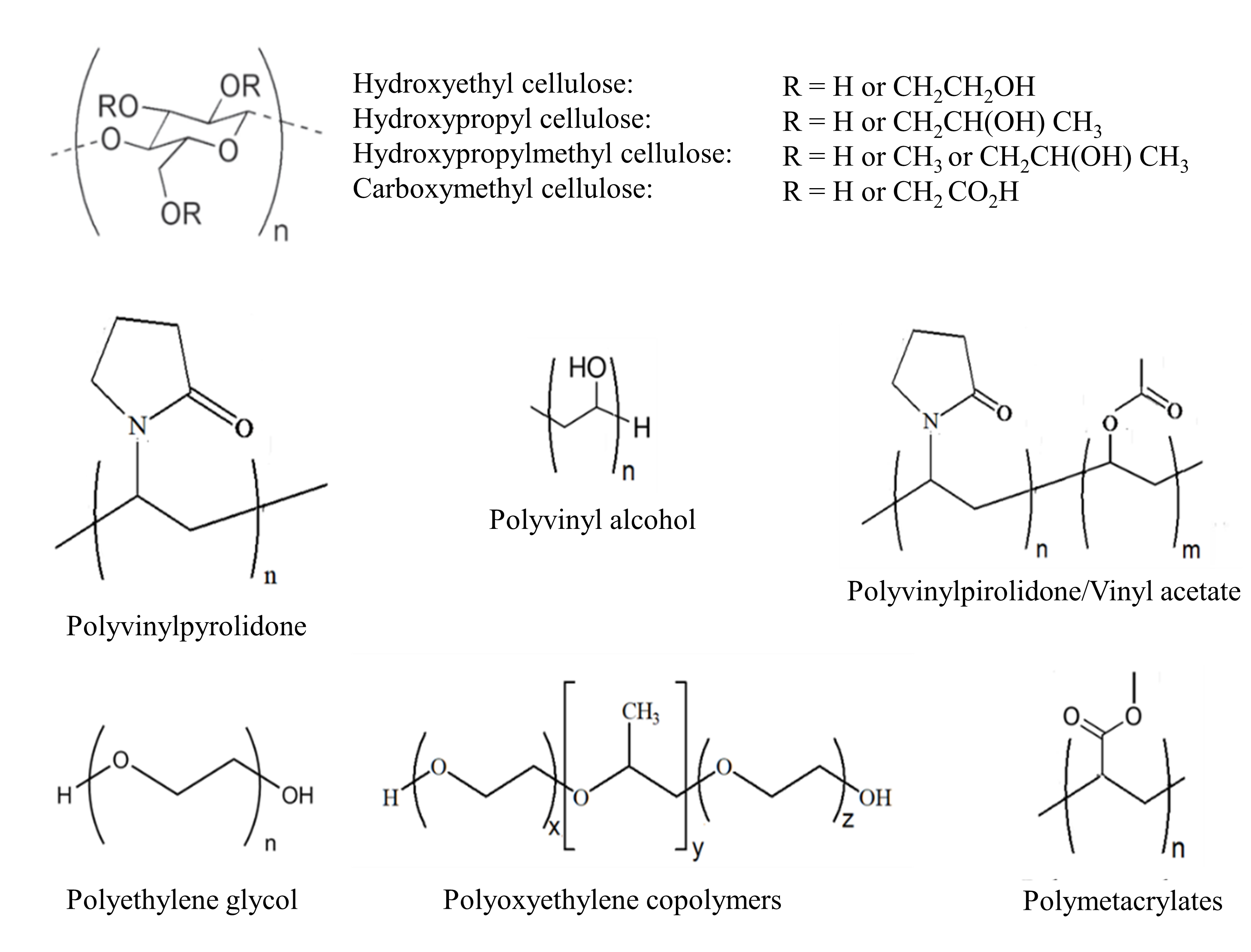
Fig. 3. Chemical structures of main polymeric precipitation inhibitors.
In order to tailor the selection of precipitation inhibiting polymer to the specific API and lipid-based formulation, a throughout understanding of their interference with these critical processes is crucial. More specifically, they influence the kinetics and/or thermodynamics of the system. The addition of the excipient can delay precipitation of the API, which is typically measured as prolonged supersaturation time and/or increased solubility of API, while the extent to which supersaturation is maintained could also be determined by the supersaturation holding capacity of the polymers introduced by Chavan et al. (53). Such effects can be achieved, for example, when the amphiphilic copolymer Soluplus® is added at a concentration above its critical micellar concentration. In this case, thermodynamic stabilization is achieved by the solubilization properties of Soluplus® linked to micelles formation. Otherwise, if the polymer effect manifests itself in a temporary inhibition of the onset of precipitation, the change is in the system kinetics. The ability of the inhibitor to kinetically stabilize the state of supersaturation is likely the result of the intermolecular interactions between the API and the polymer and/or its ability to sterically hinder the surface of the API and prevent precipitation (56, 57). By forming hydrogen bonds with API molecules, the precipitation inhibitor adsorbs onto the surface of the API and thus acts as a steric barrier to the adhesion of other API molecules. In this way, the polymer acts as a mechanical barrier that slows down the nucleation process and further nuclei growth (17).
Interestingly, in the presence of polymeric precipitation inhibitors, a change in crystal habit can occur. Gao et al. (58) reported the precipitation of AMG 517, a poorly water-soluble drug, in amorphous form in the presence of HPMC, while the use of PVP as a supersaturation stabilizer resulted in a crystalline form of the precipitate. As the amorphous state is known for rapid dissolution due to its higher-energy state, some studies focused on promoting precipitation of API in the amorphous form to allow faster dissolution of the API and thus improved oral absorption (18, 59).
Tables I and II provide a list of the polymeric excipients, their concentration (in percentages), and their in vitro performance in relation to their use as precipitation inhibitors ( i.e. prolongation of supersaturation time). In addition, Table I shows the amount of each component of liquid supersaturable LBSs in percentage (% of oil phase, % of (co)surfactant(s), and % of cosolvent), while Table II refers to the solids. In recent years, attention has undoubtedly shifted to the solidification of SMEDDS due to improved product stability, lower production costs, and better patient compliance (10, 60–63). In addition, the formulations of supersaturable solid SMEDDS offer the possibility of producing patient-friendly, multiunit dosage forms (such as granules, pellets, or mini-tablets), as they are capable of incorporating a higher amount of API due to their higher solubility. Therefore, adsorption to solid carriers, pelletization, granulation, spray drying, and hot-melt extrusion (HME) are among the most investigated technologies, yet the most favorable solidification technology is still on its way to establish itself in the pharmaceutical market (64).
Cellulose derivatives
The investigation of cellulose derivatives as precipitation inhibitors and stabilizers has been a major area of interest for formulation scientists focusing on the development of supersaturating formulations for the delivery of poorly water-soluble APIs (65). Various cellulose polymers have been used as inhibitors of API precipitation, with HPMC being the most studied. It has been shown that the extent of precipitation depends on the addition of HPMC (17). When HPMC is added in a wide concentration window from 2.5 to 25 %, it shows an efficient prolongation of supersaturation. Most frequently it is added in 5 % ( m/m), though. Compared to the formulation without HPMC, where the drug precipitated quickly (reportedly, turbidity of the dispersion medium was observed first, followed by the formation of a white solid precipitate), the addition of the polymer prolonged the drug concentration beyond equilibrium solubility by two more hours (52). Other results indicated that the presence of HPMC at 5 % ( m/m) can delay the precipitation of t-resveratrol and maintain a higher apparent concentration for about 60 min (66), longer than when added at 1 % ( m/m). Even when the polymer is not a constituent of the formulation, but is instead dissolved in the release medium at a remarkably low concentration of 0.025 % prior to the dissolution test, its presence effectively suppresses the precipitation of the poorly water-soluble drug X and maintains supersaturation for at least 6 hours (17). The comparative in vivo studies of supersaturable SEDDS and simple SEDDS clearly confirm that the presence of HPMC has a significant inhibitory effect on drug precipitation, as high concentrations of paclitaxel included in the supersaturable formulation led to a higher maximum concentration ( cmax) and increased oral bioavailability (52).
As the HPMC molecule is rich in free hydroxyl groups (hydrogen bond donor), the interaction with hydrogen bond acceptor groups is expected to increase the energy barrier for the onset of nucleation, and thereby stabilize the metastable supersaturated state. In the study of saquinavir-loaded supersaturable SMEDDS, HPMC was found to interact more strongly with the drug molecules than PVP K90, due to the formation of hydrogen bonds. Therefore, it was hypothesized to inhibit the precipitation of the API to a greater extent than povidone, resulting in higher solubility of the drug in the diluted formulation (67). Another proposed mechanism for the maintenance of supersaturation by HPMC is associated with the formation of cellulose bundles in water (17). As reported by Albaidhani et al. (68), the addition of HPMC significantly delayed the precipitation of candesartan cilexetil as the HPMC chains swelled. In addition, cellulose polymer chains can also adsorb onto the surface of nuclei or crystals and hinder the incorporation of the molecules into the crystal lattice. In this way, the polymer acts as a mechanical barrier that slows down the process of nucleation and crystal growth (17, 69).
HPMC is available in a wide range of substitution grades, on which the inhibitory effect depends (54). Different ratios of hydroxypropyl and methyl substitution give each type of hypromellose ( i.e. E, F, and K grades) its specific characteristics. The low molecular weight and low viscosity grades are most commonly used in supersaturable LBSs (E grades, e.g. E5LV, E15LV, E50LV). Due to their good water-solubility, blending in lipid formulations is a challenge. Therefore, the polymer powder is usually dispersed in simple LBS to obtain a uniform HPMC suspension, which can be classified as supersaturable LBS (52). In support of this, Suys et al. reported that HPMC E4M forms a stable suspension in which no polymer particles sink to the bottom and no sampling difficulties are encountered (54). In short, the study also showed that water-soluble polymers, when stably and homogeneously suspended, can prevent drug precipitation as effectively as those dissolved in LBFs. It is unlikely that in formulations with HPMC concentrations of 20 % or more, the liquid state could be converted to a paste-like consistency when the powder is suspended. As such, it would exhibit poor release on contact with the dissolution medium (17).
In addition to HPMC, other cellulose derivatives have also been used as precipitation inhibitors, which differ in their performance (70). When a supersaturable fenofibrate formulation was tested, HPMC and HPMC-acetate succinate were found to be more effective than HEC and cationic-HEC-ethoxylate (54). The use of HPMC-phthalate (in 10 %, m/m) and HPC (in 2.65 %) has also been reported (71). The incorporation of HPC into SEDDS resulted in an opaque suspension, as the cellulose derivative is not soluble in the oily vehicle, and therefore had to be suspended in the formulation. but didn’t impair the self-emulsifying ability, as the size of the obtained droplets did not change (72).
Moreover, the formulation of hard gelatin capsules filled with liquid SEDDS with suspended HPMC was compared with simple SEDDS encapsulated in an HPMC capsule shell. Both formulations prolonged supersaturated ezetimibe concentrations for 60 min compared to simple SNEDDS, indicating that the HPMC capsule acts similarly to HPMC suspended in liquid SNEDDS (73). Similar results were reported by Gao and Morozowich, who observed comparable behavior of both supersaturable capsules in terms of maintenance of supersaturation maintenance, which were superior to simple SNEDDS (17). Clinical evaluation in humans confirmed these results and showed better pharmacokinetic parameters ( cmax, as the maximum observed concentration, Tmax, as the time in which the maximum concentration was reached and Area Under the Curve ( AUC), as the integral of a curve illustrating the change in plasma concentration of API in a unit of time) for supersaturable SEDDS, indicating faster and more complete absorption. The advantage of HPMC capsules lies not only in their greater resistance to water migration (which is very important in terms of stability and storage of the dosage form), but also in the vegan population.
Adsorption onto solid carriers, fluid-bed, and high-shear granulation have also been investigated as solidification techniques for LBS. In the study on carvedilol-loaded SMEDDS, the effect on supersaturation was not directly evaluated as the concentration of API was not above its solubility limit. Instead, HPMC was added in the granulation dispersion as a binder, and both the SMEDDS granules and tablets produced showed complete drug release (74, 75).
As an acetate succinate derivative, HPMC–acetate succinate is also effective as a precipitation inhibitor in supersaturable formulations, although it was originally developed as an enteric coating polymer. Due to its amphiphilic nature, this HPMC analog can maintain supersaturated drug concentrations over a prolonged period of time by forming colloidal species in aqueous medium. Its hydrophobic regions can interact with the poorly water-soluble drug molecules, while its hydrophilic regions align with the water medium and stabilize the formed associate structures. Possible interactions with the API also depend on the acetyl and succinyl content, as HPMC–acetate succinate is available in three different degrees of substitution: L, M, and H. Each grade is also available as F (fine) or G (granular), depending on the particle size. Accordingly, grade H as the most hydrophobic derivative (with a high acetate: succinate ratio), has the potential for a stronger interaction with poorly water-soluble API (76). A stronger interaction could also lead to stabilization of supersaturation, and thus be superior to HPMC as well as PVP and PVP/VA (77, 78).
Unfortunately, the utility of HPMC–acetate succinate in LBSs may be limited by its poor solubility in formulations. Although some hydrophilic polymers can be suspended in lipid-based formulations, the suspension of HPMC–acetate succinate resulted in a viscous system with polymer aggregates that made homogeneous sampling difficult (54).
Vinyl derivatives
PVP, also known under the trade name Kollidon®, is a polymer often used in supersaturable SMEDDS formulations and is available in a variety of grades ( e.g. according to different K-values) (80). The K-value depends on the relative viscosity of the aqueous solution, which is positively correlated with the average molecular weight of the polymer (80). PVP K30 and K90 are the most commonly used and the best results in preventing precipitation are obtained when they are added in concentrations of 2.5 to 5 % ( m/m) (67, 72, 73). Nevertheless, even a low concentration of PVP K17 of 0.5 % was able to maintain a high concentration of indirubin in the aqueous medium, resulting in increased bioavailability in rats compared to a simple formulation without a polymeric precipitation inhibitor (78).
The use of PVP/VA has been much less reported in the literature on supersaturable formulations. Lee et al. (72) compared the supersaturable SEDDS containing PVP, PVP/VA, or HPC. Of all the formulations tested, PVP showed the best ability to prevent precipitation and maintained the drug concentration above 75 % for 6 h. The best performance of the vinyl polymer was explained by the formation of hydrogen bonds between the PVP molecule and the API. Since PVP molecules have many carbonyl groups that are hydrogen bond acceptors, they tend to interact with hydrogen bond donating groups. Cyclosporine A as a model drug is a polypeptide that contains both hydrogen bond donors and acceptors. As such, its hydrogen bond donor groups can associate with the carbonyl groups of PVP. The interaction between the polymer and the drug molecules influences the activation energy of nucleation and effectively slows down the onset of nucleation and consequently crystal growth. Therefore, PVP proves to be more effective in inhibiting the precipitation of drugs with hydrogen bond donor groups in the molecule compared to HPC as an example of hydrogen bond donors. However, the difference between the efficacy of the two formulations is sometimes difficult to measure. In a comparative study of supersaturable SMEDDS with HPMC or PVP, no noticeable difference was found in the ability of polymers to prolong supersaturation. Nevertheless, the formulations maintained the high drug concentration for 90 minutes, which was an improvement compared to simple SMEDDS (67). The effect of PVP on the crystallization process of API has also been investigated. Lindfors et al. reported the adsorption of PVP to the bicalutamide crystals formed in a supersaturated solution, creating a physical barrier that resulted in the inhibition of crystal growth (81).
In addition, Kollidon® (80) was also used in hot melt extrusion and spray drying, both of which have emerged as SMEDDS solidification techniques. However, for supersaturable formulations, research has mostly focused on simple adsorption techniques. Patki et al. used a co-processed excipient for the adsorption of liquid SNEDDS (82). The excipients, the mesoporous carrier Florite®100 and the hydrophilic polymer PVA, were co-processed in the laboratory by simple mixing in a glass mortar with a pestle and the addition of purified water. The study results showed a complete release of the API from the formulation, implying that precipitation of the API could also be prevented by the incorporation of a precipitation-inhibiting component in the co-processed excipients.
Soluplus®
Soluplus® is a copolymer with a PEG 6000 backbone (consisting of 13 % PEG 6000/ 57 % vinyl caprolactam/ 30 % vinyl acetate). The amphiphilic structure, composed of hydrophilic polyethylene glycol and hydrophobic polycaprolactam groups, implies its solubilizing ability, which acts like a surfactant and self-associates into colloidal structures when the concentration exceeds the critical micellar concentration. In this context, the addition of the optimal amount of Soluplus® is closely linked to the micelle formation as poorly water soluble drugs can be incorporated into the hydrophobic region of the formed micelles (83). In addition, the hydrophobic group of Soluplus® can also intercalate to the hydrophobic tails of surfactant, thus affecting the dispersed phase and making the interphase firmer/stronger. At the same time, the hydrophilic polyethylene groups of the inhibitor, which are present on the surface of colloidal species, can sterically stabilize the system and protect it from aggregation and (micro)emulsion collapse. Overall, the addition of the excipient stabilizes the supersaturation state by increasing the solubilization capacity.
Furthermore, its inhibitory effect can also be achieved by kinetically stabilizing the supersaturation. Two hydroxyl groups in the molecule, which behave like hydrogen-bond donors, can interact with hydrogen-bond acceptor functional groups in the drug molecule. The established hydrogen bonds increase the activation energy of nucleation and lead to a delay in the onset of nucleation and crystal growth of fenofibrate (84).
In the study with dutasteride-loaded supersaturable SEDDS, various polymers (HPMC 2910, PVP K90, Soluplus®, etc.) were tested for their precipitation-inhibiting effect. All inhibitors tested were added at the same concentration (15 %, m/m), with Soluplus® showing a superior ability to inhibit precipitation and maintain the highest drug concentration during the 2-hour dissolution test under non-sink conditions. Interestingly, the addition of Soluplus® in the increasing amounts of 15, 30, and 45 % resulted in a comparable dissolution profile of the API to the supersaturable SEDDS with 10 % Soluplus®. The combination of Soluplus® with other hydrophilic polymers, HPMC or Kollicoat® MAE (methacrylic acid ethylacrylate copolymer), was also investigated. Although a complementary precipitation inhibiting effect was expected, the combinations proved to be antagonistic compared to Soluplus® alone, as an increase in drug precipitation was observed.
Once again, Soluplus® proved to be the best choice among the precipitation inhibitors, alongside HPMC and PVP, as shown in the comparative in vitro study. The addition of 6.7 % excipient in liquid supersaturable SEDDS maintained the dissolution rate at ~85 % over 24 hours, which is consistent with the in vivo absorption study that showed significantly higher cmax and AUC values, thus facilitating the oral absorption of tacrolimus (compared to simple SMEDDS) (85).
In addition, Soluplus® can also be used to solidify supersaturable SMEDDS due to its widespread use in hot melt extrusion as well as the spray drying process. However, the precipitation-inhibiting effect of Soluplus® has so far only been investigated for supersaturable LBF produced by adsorption onto solid carriers, the simplest and most commonly used solidification technique for S(M)EDDS. In comparison to corresponding supersaturable liquid SEDDS, celecoxib-loaded solids exhibited a slower dissolution rate due to slower desorption from the solid carriers (86). In addition, the high amount of added polymer (18 %, m/m) could also contribute to a slower dissolution profile. Thickening the microenvironment of the formulation ( i.e. the dissolution media surrounding the formulation particles), increased the viscosity of the system, which contributed to lower molecular mobility (69).
Poloxamers
The poloxamer structure is composed of a central hydrophobic chain of polypropylene oxide with two hydrophilic polyoxyethylene chains on each side. These non-ionic three-block copolymerare distinguished from each other by the three digits, which are a function of the molecular mass of the polymer and the polyoxyethylene content. It is also known under the trade names Pluronic®, Kolliphor® P, Synperonic® PE, and Lutrol®.
Due to its amphiphilic nature, its surface-active properties have also been utilized in supersaturable formulations to prevent drug precipitation. Poloxamer 407 is the most commonly used copolymer, which is added to liquid LBSs in wide concentration ranges up to 18 % ( m/m) (optimally 5–10 %). In the case of the SMEDDS formulation, it was added under slight heating (~45 °C) and constant stirring until a homogeneous mixture was obtained (70).
Poloxamer 407, added at a concentration of 10 % ( m/m), was selected for the comparative study in SNEDDS. Compared to HPMC, polymethacrylate, and cyclodextrin polymers, poloxamer showed superior dissolution efficacy as it maintained supersaturation for the longest period of time. As discussed by the authors, when the copolymer was added at a concentration above the critical micellar concentration, the excess drug molecules were incorporated into the colloidal structures, thermodynamically stabilizing the state of supersaturation. In another study, the interaction between polymer and silymarin was determined by Fourier transform IR spectroscopy, and found that poloxamer 407 acted as a hydrogen bond acceptor for the (-OH) donor group of silymarin. In this way, supersaturation was successfully maintained by kinetic stabilization (70).
Moreover, poloxamer 407 acts as an inhibitor of P-glycoprotein, a drug efflux transporter located in the intestinal epithelium. Its inhibition increased the permeation rate and consequently, the oral bioavailability of APIs that are P-glycoprotein substrates (87).
In the study with valsartan-loaded SMEDDS, the addition of poloxamer 407 as a successful precipitation inhibitor at concentrations of 3–6 % ( m/m) (in liquids) and 2–3 % (in granules) resulted in comparable dissolution profiles (liquid SMEDDS vs. granules), whereas SMEDDS tablets (with 1.8 % ( m/m) poloxamer 407) showed slower dissolution, as expected (88).
Finally, spray drying technology was investigated for solid SMEDDS with poloxamer 188. The mesoporous carrier was used as an adsorbent to obtain a liquisolid system – a dry powder with 64 % liquid. At concentrations of 1.85 % ( m/m) (in liquid) and 1.18 % ( m/m) (in solid SMEDDS), it was able to keep canagliflozin in a dissolved state for up to 6 hours in both formulations tested (89).
Polymethacrylates
The addition of polymethacrylate polymers, known for their use in modified-release formulations, has also been shown to be effective in preventing the precipitation of APIs (57). Eudragit® is a trade name for cationic copolymers derived from esters of acrylic and methacrylic acids. They are broadly categorized according to the desired release profile and solubility in digestive fluids. The L, E, S, and FS grades are water-soluble copolymer variants used for pH-dependent drug release, taste masking, and moisture protection coating. In contrast, the RL and RS grades as water-insoluble Eudragit®s are used in delayed and sustained release dosage forms, while the N grade with neutral functional groups is used to produce a time-controlled drug release by pH-independent swelling (90).
In the study on supersaturable Type IV LBS, the addition of 1 % ( m/m) of different Eudragit®s enabled the kinetic stabilization of the supersaturation state. A difference in the ability to prevent precipitation was observed, as Eudragit® RL maintained supersaturation twice as long as Eudragit® L100 and Eudragit® E100 (54). This result was related to the good solubility of the RL grades in the lipid formulation, in contrast to the L and E grades, which had to be suspended in the SNEDDS tested (70). Lipid-soluble polymethacrylates allow processing into a single-phase formulation, which makes sampling much easier compared to hydrophilic polymers from this group. In addition, the immiscibility of polar excipients raises concerns regarding phase separation during storage. Furthermore, Eudragit® E100 precipitated when the formulation was tested in the fasted state simulated intestinal fluid, which was explained by electrostatic and/or hydrophobic interactions between the anionic amphiphilic bile salts and the hydrophobic backbone of the cationic copolymer (54, 91).
Other polymeric precipitation inhibitors
Some research studies report that cyclodextrins are able to prevent the precipitation of API due to their surfactant-like properties. In particular, the non-ionic hydroxypropyl-beta-cyclodextrin (HPβCD) was added to SNEDDS and studied for its ability to inhibit the crystallization of silymarin. Due to its water solubility, it was suspended in the SNEDDS at a concentration of 10 % ( m/m). The results demonstrated the potential use of cyclodextrins to maintain supersaturation, as HPβCD and sulfobutylether-beta-cyclodextrin maintained the metastable state for at least 4 hours (70). The authors suggested that cyclodextrins could thermodynamically stabilize supersaturation due to their solubilizing ability linked to the formation of complexes. They could also interact with the polar regions of the API through hydrogen bonding, analogous to kinetic stabilizers (37, 92).
Poly(propylene glycol) bis(2-aminopropyl ether) (PPGAE) was also evaluated as a precipitation inhibitor and reached the top 3 most effective precipitation inhibitors of the total 17 tested. When 1 %( m/m) was added to Type IV LBS, PPGAE prolonged the duration of fenofibrate supersaturation by 15 minutes compared to a formulation without polymer. In addition, evaluation of the in situ absorption of PPGAE showed a 4-fold increase in absorption, which was the best test performance. It was hypothesized that the amino groups of the polymers interact with the hydrogen bong acceptor groups of the API through hydrogen bonding. Moreover, positively charged PPGAE groups could also interact with the ionization sites of the API and thus prevent the association of the particles (54).
RATIONAL SELECTION OF THE PRECIPITATION INHIBITOR
The approach for rational selection of the precipitation inhibitor is usually performed for each individual formulation, as there is no single formula that could directly predict the efficacy of a particular inhibitor. Currently, a pre-screening of potential polymers is performed to find the optimal combination of API and precipitation inhibitor, as a rational and predictive selection of the candidate cannot be based solely on literature data, but should be combined with the physicochemical properties of the drug and excipient.
Precipitation inhibitors can prevent API precipitation by a number of possible mechanisms that depend on the properties of the precipitation inhibitor, the API, and the medium (37). It has been shown that the process is significantly influenced by several factors, i.e. affinity for hydrogen bonding and hydrophobic interactions between the polymer and the API, solution viscosity, steric hindrance of the molecular structure, and polymer molecular weight (55). The overall inhibitory effect is the cumulative result of the simultaneous action of different mechanisms, with the relative contribution of each mechanism determining the final outcome (38).
Hydrophobic interactions occur upon contact between the hydrophobic regions of the molecule, and higher hydrophobicity is associated with higher adsorption affinity. When comparing two HPMC grades that differ in the side-chain substitution group (but have the same viscosity), the more hydrophobic HPMC-E showed a better inhibitory effect on precipitation than HPMC-K, possibly due to a higher methoxy substitution than in the K-grades (in HPMC-E the degree of substitution of the methoxy groups is 1.9, while in HPMC-K it is only 1.4) (94).
Regarding the molecular weight of the polymers, higher molecular weight polymeric precipitation inhibitors are expected to interact strongly with the API molecules, thereby effectively maintaining supersaturation and preventing precipitation. Such excipients are also known to increase the viscosity of the solution due to lower molecular mobility. In addition, larger molecules introduce more functional groups into the system, which are also capable of forming interactions. In fact, branched polymer chains can sterically hinder precipitation nuclei and thus delay nucleation and crystal growth (13). The rigidity of the structure could also play a role, as polymers with a structure that is difficult to bend are more easily adsorbed on the crystal surface. In contrast, molecules with a flexible structure are more able to rotate, and therefore interact less strongly with the API surface (95). Since viscosity and average molecular weight also differ between HPMC grades, one might expect less viscous grades, which also have a lower molecular weight, to maintain supersaturation over a shorter period of time compared to more viscous grades. This was found to be true in the case of HPMC, where HPMC E5LV was able to delay the precipitation of ezetimibe for 90 minutes, whereas HPMC E50LV was able to maintain the same effect for twice as long (73).
Data from the literature also suggests that polymeric precipitation inhibitors need to be treated as drug-specific “tools” and that their selection needs to be evaluated on an individual basis, as the type and amount of polymer appears to be formulation-specific (54). In order to recognize the efficacy of supersaturable systems compared to simple LBSs, both formulations should be loaded with the same amount of API, and the tests should be performed under the same conditions.
In addition to in vitro supersaturation tests, the bioperformance of LBSs can be determined by in vivo studies in animals (96–99). Some supersaturable LBSs that significantly improved intestinal absorption in vivo are commented in Table I and Table II. The studies were mainly conducted on rats and rabbits, and the effect is mainly assessed by using pharmacokinetic parameters: cmax, Tmax, and AUC, presented as average values.
In summary, the strategy based on the prior review of the relevant literature sources would include different precipitation inhibitors applied in lipid formulations, where precipitation inhibition is expected to be determined by the physicochemical properties of both the drug and the inhibiting excipient in relation to their mutual specific interactions. In this context, those excipients that are expected to be effective should be included in the pre-screening studies. For further detailed screening, different concentrations of inhibitors that show a superior inhibitory effect on precipitation should be carefully investigated in order to achieve the best result. This should best be combined with quality-by-design-based development that relies on complementary data obtained through advanced analytical techniques such as NMR, IR, Raman, and fluorescence spectroscopy as well as calorimetric techniques and atomic force microscopy. In this way, better insight into the interaction mechanisms between API and polymer are given, which contributes to a better understanding of the processes involved in inhibiting precipitation. Combined with experience, this can then lead to the selection of optimal precipitation inhibitors for supersaturable formulations (43, 93).
Table I. The report of numerous examples of liquid supersaturable LBSs evaluated for supersaturation effect; PK–pharmacokinetic; PPGAE – poly(propylene glycol) bis(2-aminopropyl ether)
Table II. The report of numerous examples of solid supersaturable LBSs evaluated for supersaturation effect; PK – pharmacokinetic
CONCLUSIONS
The formulation of supersaturable LBSs is a promising strategy to improve the bioavailability and/or lower the dose of poorly water-soluble APIs. By inducing and stabilizing a supersaturation state through the use of polymeric precipitation inhibitors, intensive research to provide a controlled and robust process of transforming liquid SMEDDS to solid dosage forms is paving the way to their future technological competitiveness. The inhibitory effect of the polymers is related to several mechanisms: physicochemical properties of the inhibitor (the affinity for hydrogen bonding and hydrophobic interactions between the polymer and API), its viscosity in a solution, steric hindrance of the molecular structure and the molecular weight of the polymer. Until now, there were no supporting guidelines or polymer- related tools that could estimate the overall inhibitory effect as a cumulative result of different mechanisms, and thus assure the optimal efficacy. The combination of a solid theoretical background and practical experience remains the most straightforward selection strategy for optimal polymer choice, aiming to achieve the right balance between drug solubilization, supersaturation, and precipitation, which would lead to an overall improved absorption of the API. In line with this, the production of smaller final dosage forms could also be achieved, together with reduced irritation of the gastrointestinal tract. The latter is not only related to the administration of lower SMEDDS volume compared to conventional LBS formulations but also to the presence of polymers that support the design of formulations with a lower amount of surfactant due to their ability to prevent precipitation of API in vivo. Both aspects are particularly important in the development of dosage forms for children or the elderly, who require not only safe and efficient but also easy-to-swallow and palatable dosage forms.
Funding – The authors acknowledge the financial support from the Slovenian Research Agency research core funding No. P1-0189.
Conflict of interest – The authors declare no potential conflict of interest.
Authors contribution – Collection of the literature and conceptualization, M.K. and A.Z.P; original draft preparation, M.K; review and editing, M.G. and A.Z.P; visualization, A.Z.P; supervision, M.G., and A.Z.P. All authors have read and agreed to the published version of the manuscript.
Availability of data and materials – The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.