
INTRODUCTION
Glioblastoma multiforme (GBM), a grade IV astrocytoma, is the most common brain tumor in adults with a high degree of malignancy and a poor prognosis (median survival time 15 months, 5-year survival rate of 4–5 %) (1). Current treatment options for GBM include surgical resection, chemotherapy, immunotherapy, and alternating electric field therapy (2). Despite multidisciplinary treatment, many patients experience drug resistance leading to treatment failure and tumor recurrence (3). Therefore, it is of the utmost importance to find innovative and more effective strategies to combat GBM.
In the last few decades, natural products and their derivatives have gained significant attention in the search for effective anticancer agents due to their diverse chemical structures and potential pharmacological effects (4). Among these, harmine, a β-carboline alkaloid isolated from the seeds of Peganum harmala, has emerged as a promising candidate, demonstrating notable anticancer properties, including potent anti-GBM activity (5–7). Studies conducted in vitro have shown that harmine has a significant inhibitory effect on the proliferation and migration of GBM cells. Furthermore, in vivo experiments on orthotopic xenograft models have demonstrated that harmine is an effective inhibitor of GBM growth, providing strong evidence of its potential in the treatment of this aggressive malignancy.
The detailed mechanisms through which harmine exerts its anti-GBM effects have been elucidated, revealing its ability to target a variety of molecular targets. Notably, harmine has been shown to inhibit the focal adhesion kinase/protein kinase B (FAK/AKT) and extracellular signal-regulated kinase (ERK) signaling pathways (8, 9), key regulators of GBM cells proliferation, survival, and invasion. Additionally, harmine suppresses the activity of dual-specificity tyrosine-phosphorylated and regulated kinase 1A (DYRK1A), a protein kinase implicated in GBM pathogenesis (10, 11). Downregulation of DYRK1A not only destabilizes epidermal growth factor receptor (EGFR), but also diminishes EGFR-dependent GBM growth, culminating in reduced cell viability, enhanced apoptosis, and attenuated tumor progression in vivo (12). Finally, given the structural similarity of harmine/β-carboline derivatives to the known heat shock protein 90 (Hsp90) inhibitors (13), it is possible that their cellular target is Hsp90, a key protein in GBM cell migration and invasiveness, as well as modulation of survival and apoptosis (14).
In contrast, ferrocene, an organometallic compound characterized by high stability, remarkable redox properties, and low toxicity (15), drew the interest of medicinal chemists due to its unique ability to enhance the biological activity of other molecules (16). Ferrocene also exhibits potent antioxidant properties by effectively scavenging reactive oxygen species (ROS) and mitigating damage induced by oxidative stress (17, 18). Within the context of gliomas and GBM, oxidative stress emerges as a defining characteristic and is linked to tumor progression, therapeutic resistance, and unfavorable patient outcomes (19). Thus, ferrocene shows promise as adjunctive therapy in anti-GBM treatment.
Molecular hybridization implies the covalent linking of two or more pharmacophores/bioactive fragments from different chemical entities to create novel compounds with improved pharmacological properties (20, 21). The combination of different pharmacophores can lead to a synergistic effect and thus enhance the potency, efficacy, and/or selectivity of hybrid molecules (22, 23). In addition, hybrid molecules can exhibit a multi-target mode of action that is especially useful for treating complex diseases such as GBM, and for overcoming drug resistance.
In our previous paper, we reported the synthesis and biological activities of harmicens, harmine/β-carboline, and ferrocene hybrid compounds (24). Harmicens exhibited significant and selective antiproliferative activity against MCF-7 and HCT116 cell lines. These findings, together with the remarkable anti-GBM properties of harmine and ferrocene's antioxidant capabilities, led us to hypothesize that harmicens could act as new anti-GBM agents. Herein, we investigate the in vitro anti-GBM and antioxidant activities of harmicens, as well as the underlying mechanisms of action.
EXPERIMENTAL
Chemistry
Harmicens, harmine//β-carboline, and ferrocene hybrid compounds, were successfully prepared as previously reported by us (24).
In vitro antiproliferative activity
The experiments were carried out on U251 (human malignant glioma cell line) according to the previously published procedure (24). U251 cells were cultured as monolayer and maintained in RPMI 1640 medium supplemented with 10 % FBS, 100 U mL–1 penicillin, and 100 µg mL–1 streptomycin in a humidified atmosphere containing 5 % CO2 at 37 °C. Cells were seeded at 3000 cells per well in standard 96-well plates and allowed to attach for 24 h. The next day, the medium was aspirated, and the cells were treated with 0.1 mL of the test compounds at concentrations ranging from 0.1 to 50 μmol L–1. Working dilutions were freshly prepared on the day of the testing. Fresh growth medium was added to the untreated control cells, which were defined as 100 % viable. DMSO (0.13 %) in DMEM was considered a negative control, whereas temozolomide (TMZ) and 5-fluorouracil (5-FU) were used as positive controls. After 72 h of treatment, the medium was removed, and the cells were incubated for 1 h with 0.1 mL of MTT dissolved in serum-deprived RPMI (0.5 mg mL–1). Then, the MTT-containing medium was removed, and 0.1 mL of isopropanol was added per well to lyse the cells and dissolve the formazan. Absorbance was measured at 570 nm using a microplate reader and was directly proportional to cell viability. Each test point was performed in quadruplicate in at least two individual experiments. The IC50 values (concentration required to decrease cell viability by 50 %) were calculated by using linear regression on the sigmoidal dose-response plots and are expressed as mean ± SD.
DYRK1A inhibition
DYRK1A inhibition was investigated as previously described (10). Briefly, DYRK1A-GST, anti-GST antibody, and ATP site-directed tracer 236 were pre-combined and immediately dispensed into 384-well low-volume black round bottom polystyrene plates to give a final volume of 7.5 µL per well. For compound addition, 50 nL of each compound (10 mmol L–1 stocks in DMSO) was then added to the assay wells using a Biomek NX workstation (Beckman-Coulter) equipped with a Pintool array (VP Scientific, USA) to give the final compound concentrations of 6.7 µmol L–1. For all plates, the outer pairs of lanes in the plates contained minimum signal (6.7 μmol L–1 harmine) and maximum signal (DMSO alone) controls to establish an assay window. Plates were covered and then incubated in the dark at room temperature for 1 h before being read with a PHERAstar plate reader (BMG Labtech, USA) utilizing an HTRF module to detect absorbance at 665 nm (tracer) and 620 nm (Eu-antibody). Primary screen data were recorded, the data files exported, and then analyzed using ScreenAble (ScreenAble Inc., USA). The experiment was repeated for compounds showing greater than 50 % inhibition, by 10-point, 2-fold dose-response curves using the automated DYRK1A time-resolved fluorescence energy transfer (TR-FRET) assay. Dose-response data were loaded in ScreenAble and IC50 values were determined.
Hsp90 inhibition
The binding of selected compounds to the Hsp90β C-terminal domain (CTD) was examined using the TR-FRET kit which was acquired from BPS Bioscience (USA). The assay was performed according to the manufacturer’s instructions. In short, 5 µL of diluted Tb-labeled donor and 5 µL of diluted Dye-labeled acceptor were added to all the wells, followed by the addition of 2 µL of 100 μmol L–1 solutions of test compounds. Afterward, 5 μL of diluted PPID and 3 μL of diluted Hsp90β were added. For the positive control assay, the buffer was used instead of the inhibitor solution, while the negative control contained no Hsp90-interacting protein PPID. Both compounds and controls were prepared in duplicates and were incubated for 2 h at room temperature. Following the incubation, TR-FRET was measured using Tecan’s Spark Multimode Microplate reader (Tecan Trading AG, Switzerland). The results are presented as percentages of residual Hsp90 CTD activity which were calculated by the following equation:
%Activity = 100 × (FRETcmpd – FRETneg. ctrl)/(FRETpos. ctrl – FRETneg. ctrl)
In this formula, the FRET value is the ratio between dye-acceptor emission and Tb-donor emission.
Molecular docking studies
Harmicens 14 and 13 were docked to DYRK1A (PDB entry: 3anr) and Hsp90CTD (PDB entry: 5FWK, an adapted version according to the work of Tomašić et al. (25) was used), respectively. 3D structures of ligands were generated using IQmol and converted to pdbqt using Autodock Tools 1.5.7 (26). The latter software was also used for the preparation of receptors. Harmine (from DYRK1A), EKC20 (from Hsp90CTD), water, and any molecule that was not a fundamental part of the study were removed from receptors. Polar hydrogens and Gasteiger charges were computed and added to the protein structure which was afterwards converted to pdbqt format. The centers for the grid boxes were determined with respect to the position of the harmine (x = –7.553, y = 11.675, z = 43.539; V = 14 976 Å3) or EKC20 (x = 185.346, y = 157.419, z = 191.687; V = 20 280 Å3). Docking studies were performed by AutoDock Vina 1.1.2 (27) with the following default settings: exhaustiveness: 8, max. number of modes: 9, max. energy difference: 3. Docking poses were visualized and analyzed with PyMOL (28)/Discovery Studio (29).
DPPH radical-scavenging assay
The radical-scavenging activity against 2,2-diphenyl-1-picrylhydrazyl (DPPH) was determined according to the previously developed method (30). The reaction mixture consisted of 150 µL of the corresponding compound diluted in methanol, or 150 µL of pure methanol for estimating initial absorbance (Actr), and 150 µL of DPPH solution in methanol (0.1 mg mL–1). It was incubated for 30 min and the absorbance (A) was measured at 530 nm. The percentage of inhibition was calculated using the following formula:
Determination of total reducing power
The quantification of the total phenolic content was carried out by the Folin-Ciocalteu (F-C) method according to Singleton and Rossi (31) with some modifications (32). The reaction mixture was prepared in a 96-well plate by adding 20 µL of the diluted compound (2 mg mL–1) and 50 µL F-C reagent (10 %, V/V). The reaction mixture was shaken and incubated for 5 min at 37 °C, whereupon 160 µL 700 mmol L–1 Na2CO3 was added into each well. After incubating at 37 °C for 30 minutes, the absorbance was measured at 750 nm. The calibration curve was prepared with gallic acid and the total reducing capacity was expressed as mg of gallic acid equivalents (mg/g GAE).
EPR spectroscopy
1 mmol L–1 stock solution of DPPH in methanol was prepared and calibrated using light absorption at 515 nm. 1 mmol L–1 solutions of the compounds to be tested in methanol were freshly prepared and used immediately. EPR measurements were carried out overnight at room temperature on a Bruker Biospin ElexSys 500 continuous wave EPR spectrometer (Bruker, USA) with an ER4122SHQE cavity resonator and ER049X (SuperX) microwave bridge. A 3 mm precision NMR tube (Wilmad 327-PP-7), shortened to 12 cm, was used as a capillary holder. The typical values of the measurement parameters were as follows: microwave frequency ~9.88 GHz, microwave power 10 mW, modulation frequency 100 kHz, modulation amplitude 3 G, lock-in amplifier time constant 10.24 ms, sweep range 120 G, sweep time 41.94 s. Two sweeps were recorded at each time point of the 2D-time decay series. Two types of compounds were measured each time: 1) the baseline compound, which was a mixture of 40 μL stock solution and 120 μL methanol, and 2) the reaction mixture, which contained 40 μL stock solution, 40 μL of the drug compound, either as made or 1:1 diluted by methanol, and 80 μL of methanol. Two 50 μL capillary micropipettes (ISO7550) were loaded from each mixture. Each baseline capillary was measured once at the beginning of the experiment. Then one of the reaction mixture capillaries was measured once, and these three capillaries were kept in darkness. The second reaction mixture capillary was then loaded and recorded for a two-dimensional (2D) time decay series, which usually lasted for one and a half hours. We then repeated the single point measures of the other three capillaries once more, before loading the fourth capillary for a second 2D time decay series overnight.
Density functional theory calculations
RESULTS AND DISCUSSION
Chemistry
Harmicens represent hybrids composed of harmine/β-carboline and ferrocene moieties covalently bound via a 1H-1,2,3-triazole or an amide, leading to triazole- (TT, 1, 2, 5, 6, 9–12, 15, 16) or amide-type (AT, 3, 4, 7, 8, 13, 14, 17, 18) compounds (24). Structural diversity was increased by the preparation of hybrids in five positions of the β-carboline ring (C-1, C-3, O-6, O-7, and N-9) and the presence of a methylene bridge in the linker between the triazole/amide and ferrocene (Fig. 1). Cu(I)-catalyzed azide-alkyne cycloaddition was the method of choice to prepare TT harmicens, whereas synthesis of AT harmicens was accomplished using standard coupling reaction.
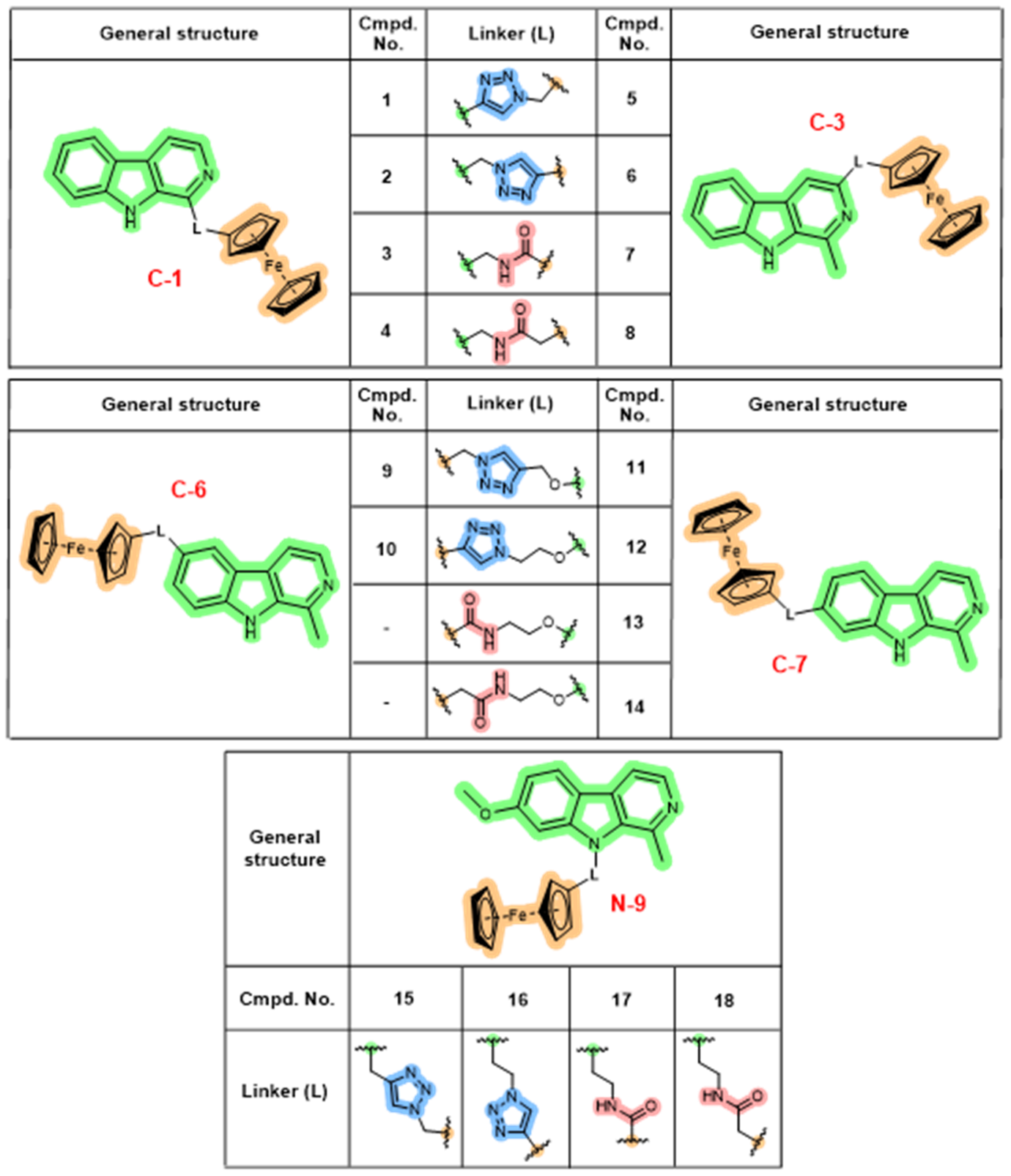
Fig. 1. Harmicens – harmine-ferrocene hybrid molecules (harmine/β-carboline is marked in green, ferrocene in orange, triazole in blue, and amide in red).
In vitro antiproliferative activity
The effect of harmicens 1–18 against human malignant glioma cell line (U251) was examined in vitro using MTT assay and the obtained results are given in Table I. The parent compounds harmine and ferrocene, as well as the reference drugs 5-FU and TMZ, were used as positive controls. Initially, we conducted a preliminary screening, and only compounds that caused a reduction of mitochondrial metabolic activity by over 50 % at a concentration of 50 μmol L–1 were selected for subsequent determination of IC50. To determine harmicens’ selectivity, we compared their effects against U251 with that on the human embryonic kidney cell line (Hek293, non-cancer cell line), investigated in our earlier work (24). Selectivity indices were calculated as ratio IC50(Hek293)/IC50(U251).
Harmicens showed remarkable anti-GBM activity (5/18 hybrids exerted low micromolar IC50 values), which was stronger than that of the reference drug TMZ. In general, TT harmicens were more active than their AT counterparts (4/5 compounds with single-digit micromolar IC50 value belong to the TT class). The most active and selective compounds were TT harmicenes 5, prepared in the C-3 position of the β-carboline ring (IC50 = 3.7 ± 0.1 μmol L–1, SI = 12.6), and 9 (IC50 = 7.4 ± 0.5 μmol L–1, SI = 5.8). Importantly, both compounds exerted two orders of magnitude stronger antiproliferative activity than the reference drug TMZ and were significantly more selective than 5-FU (SI = 1.1). Harmicens 10, 12, and 14 showed strong, but non-selective antiproliferative activity and significantly affected Hek293, while antiproliferative activities of compounds 6, 13, and 18 were moderate.
SAR analysis revealed that antiproliferative activity was influenced by the position of the linker on the β-carboline core. In the TT group of harmicens, the introduction of the linker at the C-6 position was the most preferable for antiproliferative activity. In contrast, among AT harmicens C-7-tethered derivatives exerted the strongest antiproliferative activity. In both TT and AT groups of harmicens, C-1 and N-9 tethered compounds were less active, suggesting that these positions are unfavorable for the introduction of the linker. We also noticed that harmicens’ selectivity depended on the structure of the linker. Triazoles 5 and 9, with a methylene group incorporated between the triazole and ferrocene rings, demonstrated selective activity, while their counterpart compound 10 did not. This emphasizes the importance of the methylene spacer for selectivity. Interestingly, this trend appears to be less pronounced among AT harmicens.
Table I. Antiproliferative activity and DYRK1A inhibition of harmicens 1–18
aIC50 – the concentration that causes 50 % (growth) inhibition; b SI – selectivity index; c weak or no inhibition at the highest tested concentration; d n.a. – not applicable; e n.d., not determined; f HAR – harmine; g FER – ferrocene; h 5-FU – 5-fluorouracil; i TMZ – temozolomide.
In vitro DYRK1A inhibition
To gain insight into the potential mechanism underlying the anti-GBM activity of harmicens, we conducted an in vitro assay to evaluate their inhibitory effects on DYRK1A, a kinase known to play a crucial role in the complex pathophysiology of GBM. In our investigations, an automated one-step TR-FRET assay for DYRK1A using the LanthaScreen® Eu Kinase Binding Assay Technology was employed to identify competitive kinase inhibitors by displacement of a fluorescently-labeled ATP site-directed tracer, which fluoresces only when proximal to a fluorescently-labeled antibody that recognizes a tag also present on the DYRK1A (41, 42). Results are given in Table I.
Amides 14 and 18 exhibited pronounced inhibition of DYRK1A, with IC50 values in the submicromolar range. Likewise, triazoles 12 and 15 displayed considerable activity, albeit with IC50 values in the micromolar range. However, none of the other compounds inhibited DYRK1A, even at the highest concentration tested. We found a correlation between the antiproliferative activity of specific harmicens (12, 14, and 18) and their ability to inhibit DYRK1A, suggesting that DYRK1A inhibition could be their potential mechanism of action.
In vitro Hsp90 inhibition
The capacity of harmicens to inhibit Hsp90 CTD was investigated using a commercially available TR-FRET assay kit. This assay is based on the disruption of the interaction between Hsp90β's CTD and its cochaperone cyclophilin D (PPID) using Hsp90 CTD inhibitors. When the donor beads, which are attached to PPID via a GST-tag, are excited, they transfer energy to the acceptor beads (attached to Hsp90 via biotin). However, if the interaction is disrupted by Hsp90 CTD inhibitors, the energy transfer is prevented, resulting in a measurable readout for the inhibition of Hsp90 CTD (43).
Six compounds with significant antiproliferative activities were tested for Hsp90 CTD inhibition at 100 μmol L–1 concentrations. Results are expressed as Hsp90 CTD residual activity and graphically shown in Fig. 2. Compound 13 was found to be the most effective inhibitor of Hsp90CTD, reducing its residual activity to 49.9 %. Compounds 12 and 14 exhibited inhibitory effects, but their potency was comparatively lower than that of compound 13.
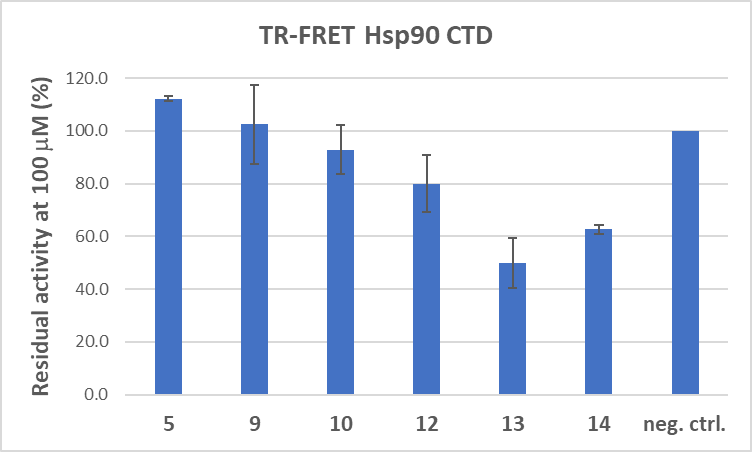
Fig. 2. Biochemical evaluation of Hsp90β CTD binding using TR-FRET.
Molecular docking studies
Molecular docking analysis was employed to offer more insight into the binding mode of the most potent compounds 14 and 13 towards DYRK1A and Hsp90CTD, respectively. To accomplish this, we utilized the crystal structures of human DYRK1A in complex with harmine (PDB code: 3ANR), while compound 13 was docked to the allosteric binding site of the Hsp90 CTD identified by Tomasic and coworkers (25). Molecular docking analysis was performed using AutoDock Vina and the results were analyzed in PyMol/Discovery Studio.
Harmicene 14 showed a remarkable binding affinity of –10.8 kcal mol–1 for DYRK1A, underlining its significant interaction with the protein. Within the binding pocket, it formed two hydrogen bonds: first, between the nitrogen atom at position 2 of the β-carboline core and LYS188, and second, between the oxygen atom at position 7 and Leu241, as shown in Fig. 3a. In addition, harmicene 14 established hydrophobic interactions within the binding site. These include π-alkyl interactions with Leu294, Val222, Lys188, Val306, Val173, Leu241 and Ala186, while hydrophobic alkyl interactions were observed with Val173 and Lys188.
The binding affinity of harmicene 13 for Hsp90βCTD was determined to be –9.8 kcal mol–1. The amino group at the N-9 position of the β-carboline ring formed a hydrogen bond with GLN493, while the NH group of the amide bond formed a similar interaction with GLY667, as depicted in Fig. 3b. The methyl group at C-1 of the β-carboline core showed hydrophobic alkyl interactions with ILE486 and VAL494. Furthermore, the β-carboline ring contributes to a π-alkyl interaction with the ALA600, while ferrocene is involved in a π-alkyl interaction with PRO673.
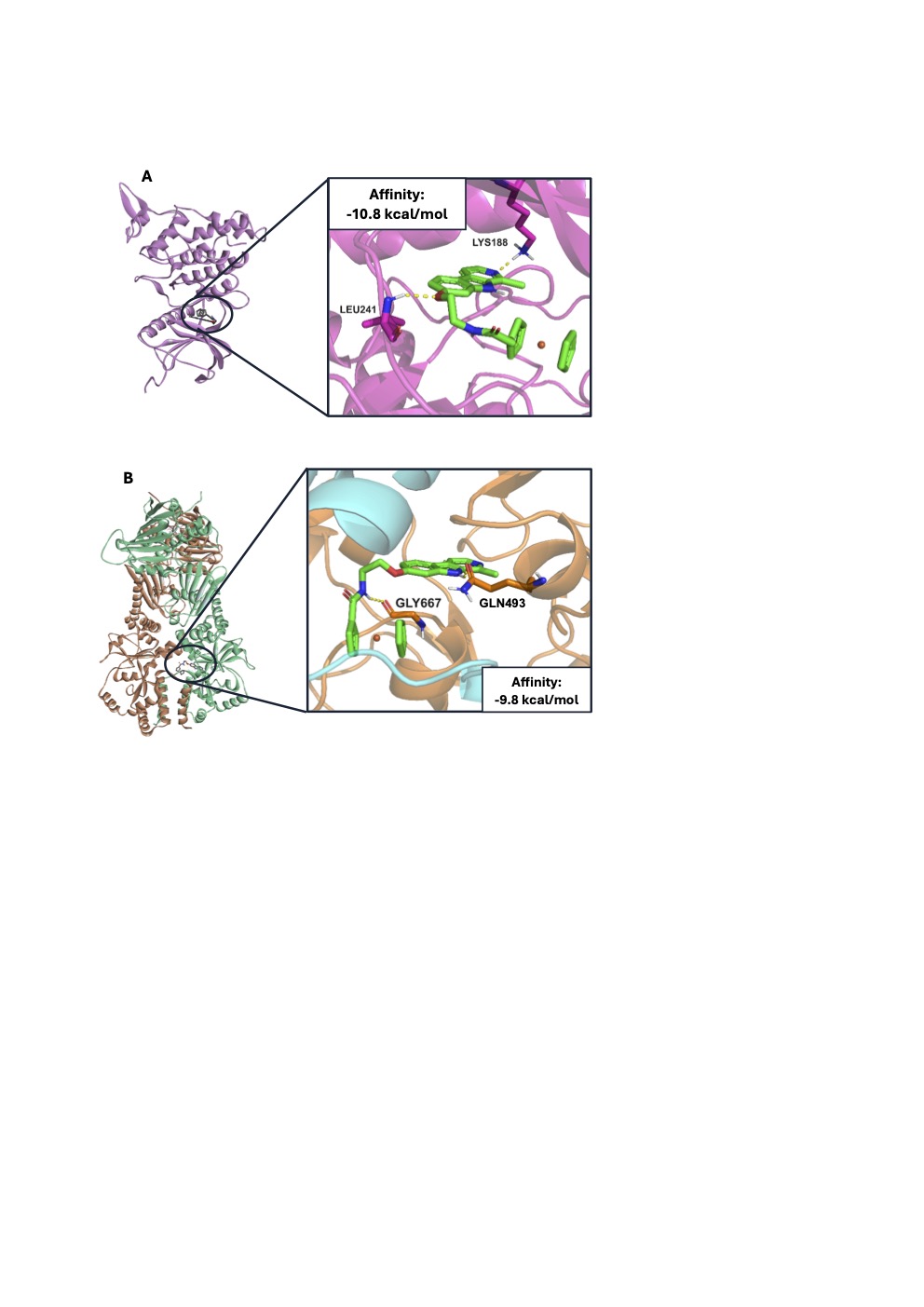
Fig. 3. a) Docking binding modes of compound 14 (green) in the DYRK1A (rose) and b) compound 13 (green) in the Hsp90β CTD (protomers are coloured dark orange and cyan, respectively). For clarity, only the hydrogen bonds (dashed yellow lines) and interacting amino acids are shown.
Antioxidant activity
Since ferrocene possesses significant antioxidant properties, which could be particularly advantageous in treating GBM, we aimed to explore the antioxidative potential of harmicens spectrophotometrically using two methods: the DPPH radical-scavenging assay and the Folin-Ciocalteu method.
In the DPPH assay, an antioxidant transfers an electron to DPPH, causing the original purple color of DPPH to fade, turning either colorless or pale yellow. This color change can be measured by spectrophotometry and is directly correlated to the antioxidant capacity of the tested compound: the more pronounced the color change, the stronger the antioxidant potency. On the other hand, the Folin-Ciocalteu method employs the reduction of phosphotungstic-phosphomolybdic acids, known as Folin-Ciocalteu reagent, by antioxidants under alkaline conditions. This reduction results in a blue-colored complex, the intensity of which directly correlates with the total antioxidative capacity of the compound.
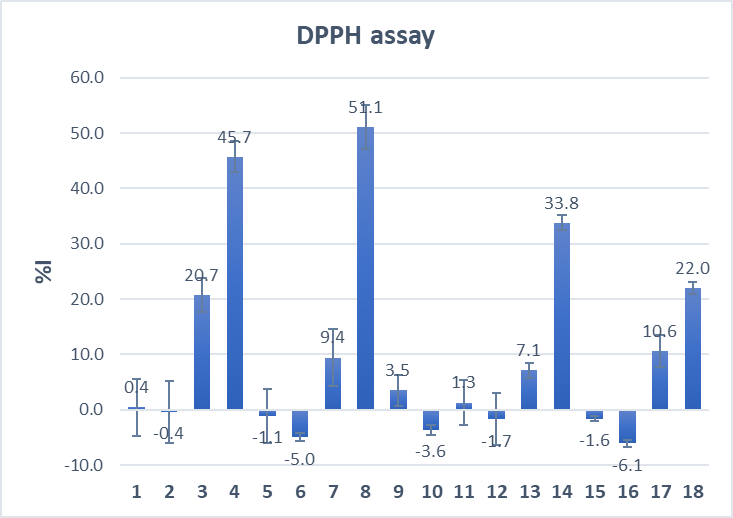
In both assays, compounds 4, 8, 14, and 18, amides with methylene spacer between ferrocene and amide group, showed the strongest antioxidative properties (Fig. 4). Latter indicates that the type and structure of the linker, but not the position of the linker at β-carboline core, determine the antioxidative properties of harmicens.
a)
b)
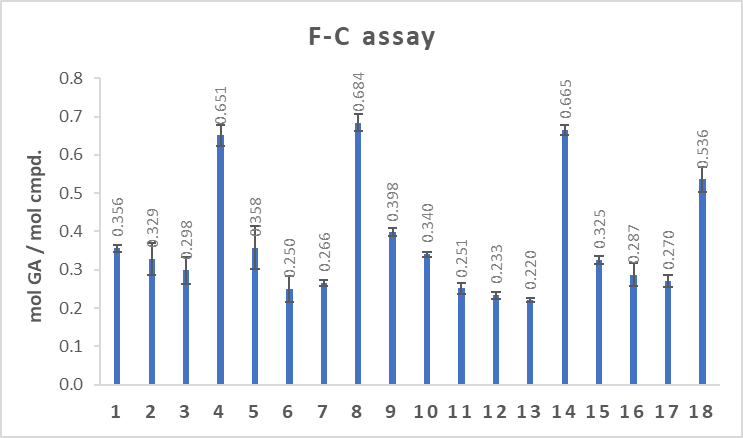
Fig. 4. Antioxidant activity of harmicens determined by: a) DPPH radical-scavenging assay and b) Folin-Ciocalteu method.
EPR spectroscopy
Electron paramagnetic resonance (EPR) was used as a second and independent method to verify the antioxidant activity determined previously by spectrophotometric methods. We discovered from the literature (44–46), and some preliminary tests that the compounds we were studying could have exhibited different DPPH scavenging assay values due to varying scavenging rate differences. To get a more accurate understanding of the scavenging rates, we decided to measure the DPPH signal decay induced by harmicens (5, 9, 12, 13, 14, 18) by EPR spectroscopy overnight.
Two quantities derived from the spectra collected were used to quantify the remaining DPPH. One was the peak-to-peak amplitude of each EPR spectrum. The other was the area under the resonance absorption peaks (so-called “double integral” in EPR spectroscopy). We chose the latter one to present the results, because it was found to be more consistent than measuring peak-to-peak amplitudes, despite being less straightforward. Two compounds (14 and 18) managed to scavenge all DPPH in the 1:1 mixture overnight. 2:1 reaction mixture of these two compounds was subsequently measured and found to have nearly all DPPH scavenged as well (Fig. 5). We therefore estimated the scavenging capacity of these two compounds to be approximately one (compound) to two (DPPH). Compound 14 scavenged DPPH slightly faster than harmicene 18. The scavenging rates of these two compounds were considerably slower than the antioxidants used in usual applications (44–46). The other four compounds did not finish scavenging all DPPH in the 1:1 mixture overnight. Their 2:1 mixture was not made because the reaction would have taken too long time to finish.
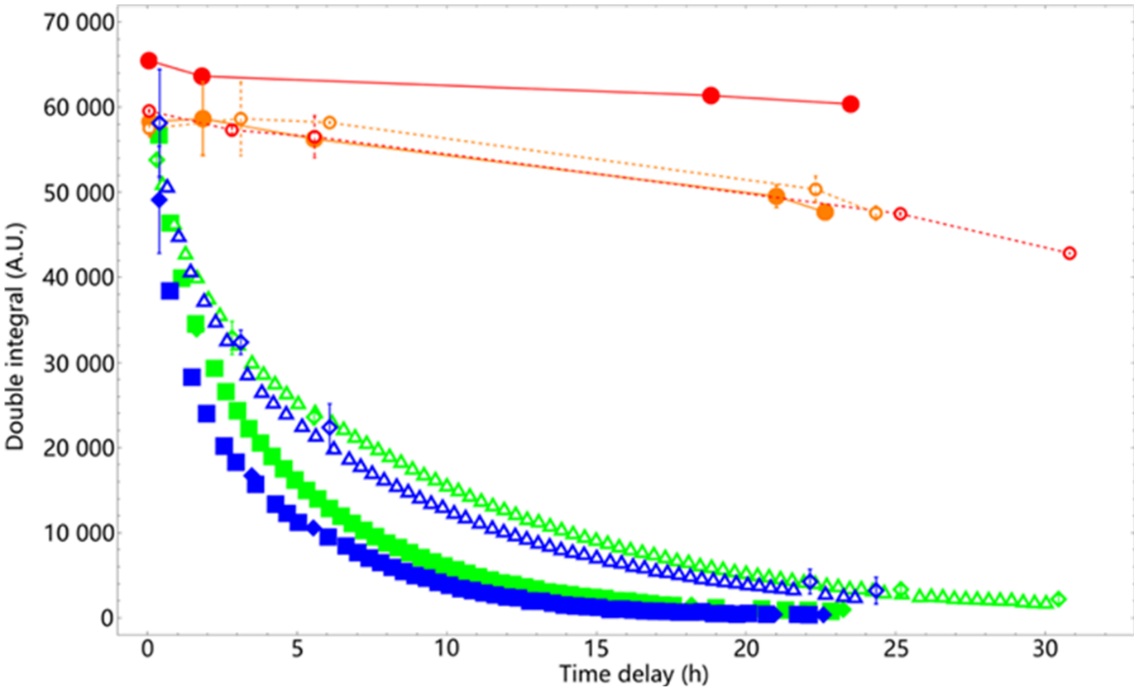
Fig. 5. The DPPH double integral value reduction results of overnight measurements of sample 14 (blue), and sample 18 (green). The corresponding baseline measurements are marked red (14) and orange (18). The solid/open blue markers are from the 2D time decay measurement of the 1:1/2:1 mixture of DPPH and sample 14. The solid/open green markers are from the 2D time decay measurement of the 1:1/2:1 mixture of DPPH and sample 18. The blue/green markers with error bars are averages and standard deviations of the immediate subsequent measurements of two capillaries loaded with the same reaction mixtures, respectively. The red solid/open markers with error bars linked by red solid/dashed lines are baseline measurements (averages and standard deviations) for the 1:1/2:1 mixture of DPPH and sample 14. The orange solid/open markers with error bars and linked by orange solid/dashed lines are baseline measurements (averages and standard deviations) for the 1:1/2:1 mixture of DPPH and sample 18.
DPPH double integral time decay records of all six compounds are plotted in Fig. 6. We observed that harmicens 5 and 9 scavenged large proportions of the DPPH signal after one night, but not all of it, while harmicens 12 and 13 only scavenged small parts of the DPPH signal overnight.
The slow scavenging rates of all compounds measured by EPR could explain the small assay values obtained by spectrophotometry, as the latter were measured after a short and fixed time period (half an hour). Slower compounds would induce fewer DPPH signal reductions and thus smaller assay values. The fastest scavengers in the EPR experiment, harmicens 14 and 18, did result in the largest assay values in the spectrophotometry experiment. On the other hand, the slowest scavengers, harmicens 12 and 13, in the EPR experiment had larger assay values than harmicens 5 and 9 in the spectrophotometry experiment. The exact reason for this difference is still unclear.
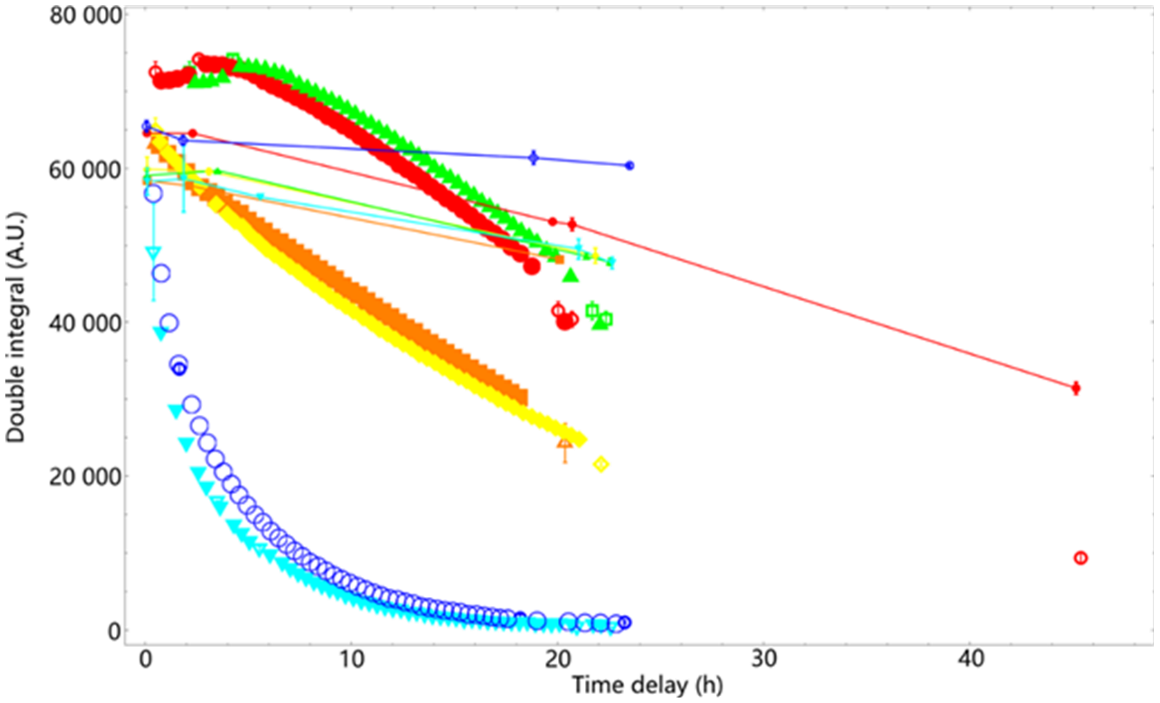
Fig. 6. The DPPH double integral value reduction results of overnight measurements of a 1:1 mixture with harmicens 5 (orange), 9 (yellow), 12 (red), 13 (green), 14 (cyan), and 18 (blue). Solid lines connecting small symbols present the baseline measurements for the reaction mixture of the same color, respectively.
Density functional theory calculations
As stated above, harmicens 14 and 18 have the fastest scavenging rate among compounds. To properly describe which part of the structure is responsible for the fast scavenging, an in-depth analysis of radical stabilities was performed. First, relative redox potentials were calculated between ferrocene and harmicens, and then bond dissociation energies (BDEs) for hydrogen atom transfer reactions were estimated, through isodesmic reactions, using a previously published approach (47, 48).
As seen in Fig. 7, amide directly connected to the ferrocene moiety has the lowest potential, relative to ferrocene, while triazole directly connected to the ferrocene, either via N- or C- has more 800 mV positive shift compared to ferrocene. Those two combinations are present in structures 4 and 8, but their differences in the potentials have not been translated into differences in biological activity. Therefore, scavenging has to be observed through differences in the BDE of different fragments. All relevant combinations of ferrocene and linkers vs. harmicene and linkers are shown in Fig. 7. It is important to note that all ferrocene-containing structures are less stable than referent system methane/methyl radical. Linkers by themselves also produce radicals that are more reactive than the methane/methyl radical pair and ammonia/aminyl radical pair. When connected to the ferrocene, the stabilities of radicals increased, making them less reactive. Compounds 14 and 18 both have ferrocene-CH2-CONH-CH3 substructure, which is indeed the most unstable and, thus most reactive radical species. It can be postulated that this is the structural parameter responsible to the fast radical scavenging properties.
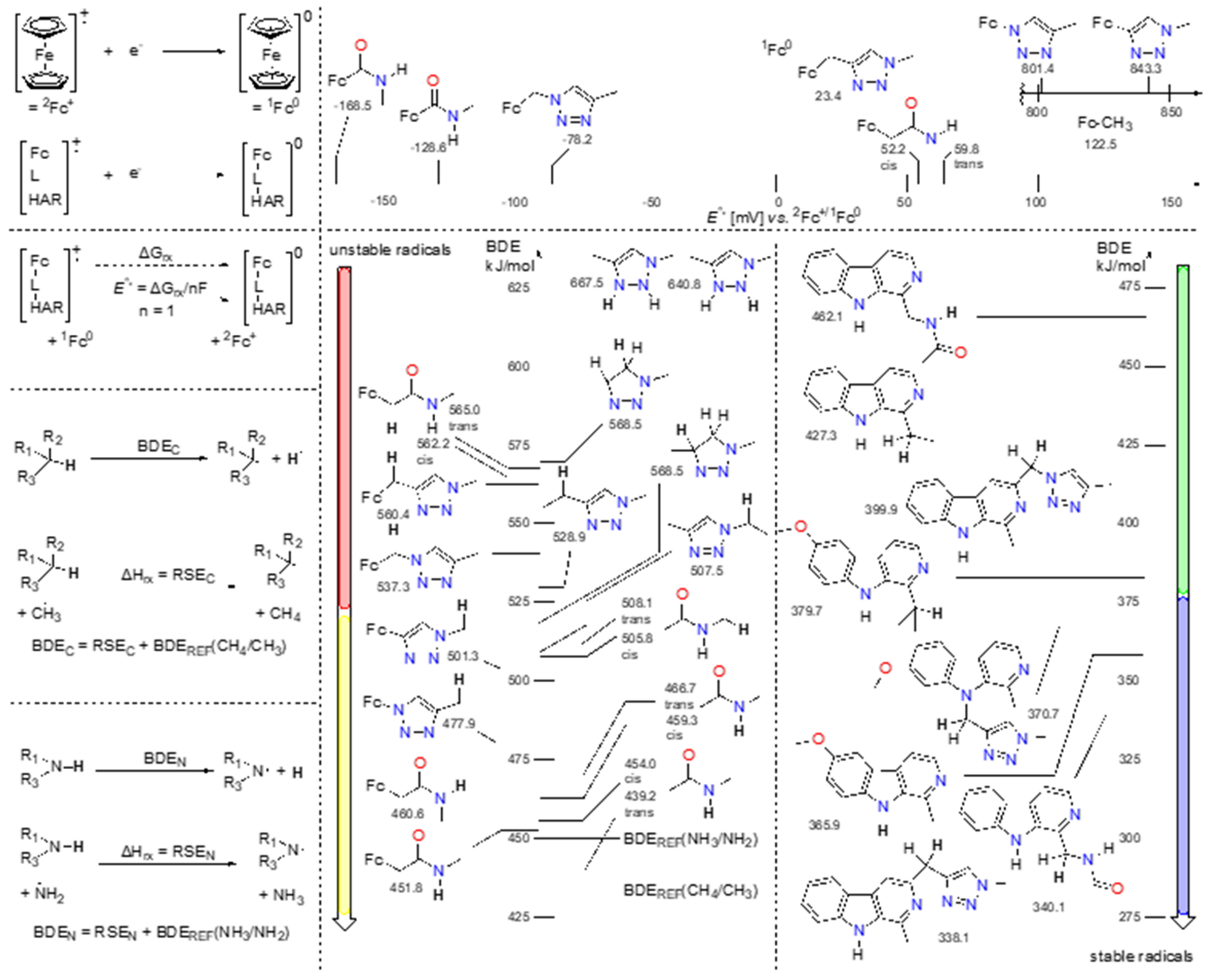
Fig. 7. Predicted electrochemical potential and bond-dissociation energies (BDEs). In the left panels are defined isodesmic equations used for the calculation of parameters.
CONCLUSIONS
Despite significant efforts, prognosis and treatment options for GBM remain poor. Thus, we decided to investigate the antiproliferative activity of a series of harmicens 1–18, harmine/β-carboline, and ferrocene hybrids, against the human malignant glioma cell line and the underlying mechanisms of action. Screening of their antiproliferative activity in vitro identified harmicens with powerful antiglioblastoma activity, which was stronger and more selective than that of the reference drugs TMZ and 5-FU. The most active and selective compounds were TT harmicens 5 (IC50 = 3.7 ± 0.1 μmol L–1, SI = 12.6) and 9 (IC50 = 7.4 ± 0.5 μmol L–1, SI = 5.8), whereas AT harmicens 10, 12 and 14 exhibited strong, but non-selective activity, in the low micromolar range. Compounds 14 and 18 demonstrated a multimodal mechanism of action, which included inhibition of DYRK1A and Hsp90 CTD, accompanied by antioxidative activity. On the other hand, the most active and selective compounds 5 and 9 were not active in those assays, implying that these compounds could be targeting alternative pathways or targets contributing to their efficacy. In our future studies, we will focus on the optimization of the structures of the most active harmicens by introducing substituents at the β-carboline core, varying the length of the linker between two moieties, and on the deduction of their mechanism of action.
Abbreviations, acronyms, symbols. – AKT, protein kinase B; AT, amide-type; BDE, bond dissociation energy; CTD, C-terminal domain; DPPH, 2,2-diphenyl-1-picrylhydrazyl; DYRK1A, dual specificity tyrosine-phosphorylated and regulated kinase 1A; EGFR, epidermal growth factor receptor; EPR, electron paramagnetic resonance; ERK, extracellular signal-regulated kinase, FAK, focal adhesion kinase; FBS, fetal bovine serum; F-C, Folin-Ciocalteu; FER, ferrocene; 5-FU, 5-fluorouracil; GAE, gallic acid equivalents; GBM, glioblastoma multiforme, HAR, harmine; HCT 116, human colorectal carcinoma cell line; Hek293, human embryonic kidney cell line; Hsp90, heat shock protein 90; IC50, the concentration of the tested compound necessary for 50 % growth inhibition; MCF-7, human breast adenocarcinoma cell line; MTT, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide; ROS, reactive oxygen species; RPMI, Roswell Park Memorial Institute; SI, selectivity index; TR-FRET, time-resolved fluorescence resonance energy transfer; TT, triazole-type; TZM, temozolomide; U251, human malignant glioma cell line.
Acknowledgments. – The authors would like to thank the Zagreb University Computing Centre (SRCE) for granting computational resources on the Supek cluster, funded by EU (KK.01.1.1.08.0001), within OPCC for Republic of Croatia.
Funding. – The work was supported by the Croatian Science Foundation (research project UIP-2017-05-5160), the project FarmInova (KK.01.1.1.02.0021), funded by the European Regional Development Fund and project Evaluation of harmine analogues as potential Hsp90 inhibitors against pediatric sarcomas, funded by the Ministry of Science, Education and Youth of the Republic of Croatia.
Authors contributions. – Conceptualization, Z.R.; methodology, G.P., D.Š., M.M., J.Y., M.T, K.W., N.G., J.D. and T.T.; analysis Z.R. and E.B.; writing, original draft preparation, Z.R., D.Š., G.P. and E.B.; writing, review and editing, Z.R. All authors have read and agreed to the published version of the manuscript.