
INTRODUCTION
Late-stage functionalisation (LSF) has emerged as a transformative strategy in medicinal chemistry, providing powerful means to generate new potential drugs from existing lead compounds. This approach involves the modification of complex molecules at a late synthetic stage, allowing for the introduction of diverse functional groups without the need for extensive synthetic reroutes. By enabling the fine-tuning of pharmacological properties such as potency, selectivity, and metabolic stability, LSF plays a crucial role in optimising lead compounds and accelerating the drug development process (1, 2).
Among the "reaction toolbox" for LSF are ring-expansion reactions, which are particularly valuable for synthesising medium-sized and macrocyclic rings (3). These large-ring frameworks are critical in medicinal chemistry, with many drugs based on these structures on the market. The ability to create and modify these ring systems through LSF enables the development of drug candidates with unique structural and functional properties (4).
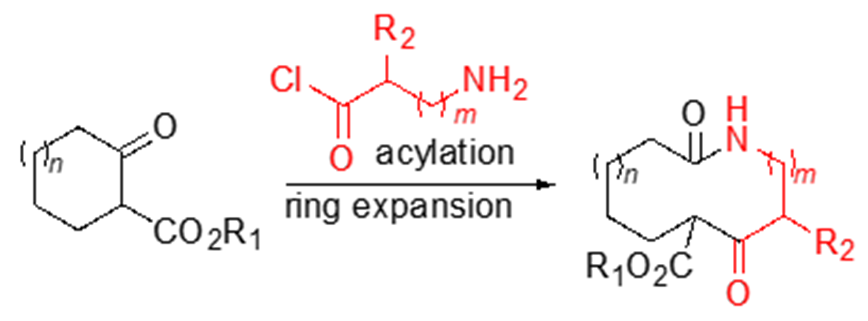
Scheme 1. Example of ring expansion in the late-stage functionalisation protocols. Adapted from reference (3).
Among macrocyclic drugs, a special place is reserved for lactams, as some of the most significant heterocycles in a variety of natural products and drugs, including antimicrobial drugs. Lactams as cyclic amides are classified by ring size, including four-membered (β-lactams, 2-azetidinones), five-membered (γ-lactams, 2-pyrrolidinones), six-membered (δ-lactams, 2-piperidinones), seven-membered (ε-lactams, 2-azepanones), medium-sized (8–11-membered), and macrocyclic (≥ 12-membered) rings. Moreover, lactams serve as conformationally restricted scaffolds that enhance the potency, selectivity, and metabolic stability of peptide-based drugs. They exhibit a broad spectrum of biological activities, making them valuable in the treatment of cancer, diabetes, infectious diseases, and more (5).
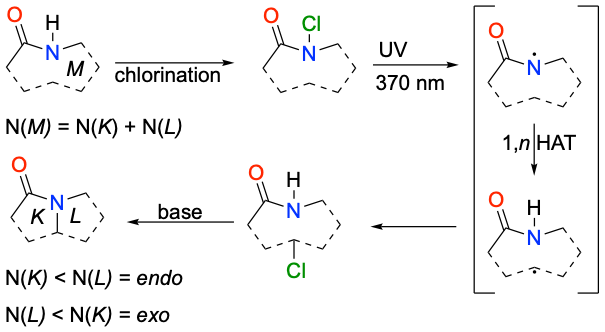
Scheme 2. Example of ring-contraction using Hofmann-Löffler-Freytag reaction.
Changing macrocyclic lactams to bicyclic lactams with smaller ring motifs, such as β-, γ-, δ, and ε-lactams, may provide new opportunities for drug development. To make a ring contraction, a previously unfunctionalized C–H bond has to be activated to form a novel C–N bond (6). The photocatalytic Hofmann-Löffler-Freytag (HLF) reaction (7–11) is a promising method for achieving this transformation and has been used in alkaloid synthesis (12, 13). It employs N-centered radicals to functionalise remote C–H bonds. Our recent investigations into the mechanistic details of the HLF reaction have provided insights into its utility in synthesising nicotine derivatives in a cost-effective way while conserving stereoconfiguration (14). The cause of regioselectivity in the HLF reaction, where more pyrrolidine than piperidine products are formed, has recently been explained, and that could steer in producing only select bicyclic structures in the macrocycle (15).
In this study, we aim to modify laurolactam (azacyclotridecan-2-one, dodecalactam), which is industrially utilised as a monomer in the polymerisation of nylon-12, to serve as a model compound and potential scaffold for further bicyclic lactam synthesis. By leveraging the HLF reaction, we intend to demonstrate the feasibility of this approach, paving the way for the development of new, stable bicyclic drug candidates from macrocyclic leads.
EXPERIMENTAL
General
The purchased compounds were sourced from Sigma-Aldrich (USA) (azacyclotridecan-2-one, trichloroisocyanuric acid (TCICA), Celite® S), Fisher Scientific (USA) (dichloromethane, DCM) and Kemika (Croatia) (sodium hydroxide). All chemicals and solvents were obtained commercially and used without further purification unless otherwise noted.
Reactions were routinely checked by thin-layer chromatography (TLC) with Merck silica gel 60F-254 glass plates (Merck, Germany) using cyclohexane/ethyl acetate/methanol 3:1:0.5 as a solvent system. Spots were visualized by spraying the TLC plates with an ethanolic solution of phosphomolybdic acid followed by heating.
Photocatalysed reactions were performed in situ (reaction of 1-Cl and EPR studies) and off-site (NMR) with Kessil PR-160L 370± 10 nm gen-2 LED UV (DiCon, USA), with an average intensity of 137 mW cm–2 when the sample is at a 6-cm distance from the lamp, according to the manufacturer (16).
Syntheses
1-Chloroazacyclotridecan-2-one (1-Cl)
TCICA (0.561 g, 2.414 mmol) was added to a stirred solution of azacyclotridecan-2-one (1) (0.433 g, 2.194 mmol) in DCM (17 mL) at 0 °C. The reaction mixture was stirred overnight at room temperature. The mixture was filtered through Celite® and the filtrate evaporated under reduced pressure to give the N-chloro derivative (0.488 g, 96 %).
Cyclisation
After one-hour long irradiation of 1-Cl using a 370 nm lamp in the round-bottom flask, while checking the reaction progress with TLC, the reaction was quenched with an excess of 0.1 mol L–1 solution of NaOH. Products were filtrate evaporated under reduced pressure and washed with deionised water.
NMR and EPR reaction monitoring
NMR spectra of the reaction mixture were obtained on a Varian Inova 400 NMR spectrometer (Varian, USA) operating at 399.90 MHz for 1H NMR and 100.6 MHz for 13C NMR. Chemical shifts (δ) are reported in parts per million (ppm). First, 1H and APT-13C spectra of the starting compound 1-Cl were acquired. Then the sample was irradiated off-site for 5 minutes from the bottom of the NMR tube using the UV lamp at 25 % power. Reaction progress was monitored with 1H spectra, and for every three irradiation intervals, APT-13C was recorded. Deuterated chloroform was used as a solvent, with some precipitation forming during the reaction. Quenched and washed products were analysed in DMSO-d6 solvent. The spectra were processed in the MNova 11.0.4 software (Mestrelab, Spain) (17) and using the NMRium online platform (Zakodium, Switzerland) (18).
EPR spectroscopy was done using a Bruker E500 ELEXSYS EPR spectrometer (Bruker, USA) with an ER4122SHQE cavity resonator. As this cavity resonator does not have an optical window for illumination, the light source was mounted underneath the cavity, with light coming through the bottom of the EPR 4 mm-inner-diameter tube, with toluene as a solvent. EPR deconvolution and simulation were done using an EasySpin module (19) with the MATLAB program package. (MATLAB, Natick, USA) EPR visualization and simulations were done using the VisualEPR Web page (20).
Computational methods
Initial structures were optimised using the semi-empirical tight-binding quantum chemical method xtb program package (21). Conformational space for reactants, products, and transition states was sampled and investigated using the Conformer-Rotamer Ensemble Sampling Tool – CREST (22) coupled with meta-dynamic simulations (23) on the GFN2-xtb level of theory (24). Selected 50 lowest energy conformations were re-optimised using density functional theory on B3LYP/6-31G(d) level of theory (25, 26) using Gaussian 16 program package (27). For each structure, frequency calculation was performed to identify if the structure is a minimum or a saddle point (minimum in all directions except in one path, identifying transition-state structure) on the potential energy surface. From the transition-state structure, an intrinsic reaction coordinate search was performed to characterise the corresponding reaction and product complexes/reactive conformers. All conformer ensembles were sorted via the CREGEN procedure, and improved energetics were obtained via single-point calculations using RO-B2PLYP-D3/G3MP2Large level of theory (28–31). Calculations were performed using the advanced computing service (cluster Supek) provided by the University of Zagreb University Computing Centre – SRCE (32) and the computational resources of the PharmInova project (sw.pharma.hr) at the University of Zagreb Faculty of Pharmacy and Biochemistry (33), and all visualisations were done using IQmol (34).
RESULTS AND DISCUSSION
HLF reaction is governed by the thermodynamic factors, namely the difference in the radical stability between reactants and products in the rate-determining step (35). A well-proven method used for the calculation and prediction of radical stabilities among the N-centered and C-centered radicals is done via modelling of isodesmic reactions involving one experimentally well-defined standard. For C-centered radicals, the standard is methane/methyl radical, while for N-centered radicals ammonia/aminyl radical is used as an anchor point, with bond-dissociation energies (BDEs) of 439.3 ± 0.4 kJ mol–1 and 450.1 ± 0.24 kJ mol–1, respectively (36). Reaction enthalpy of such isodesmic reactions is called radical stabilisation energy (RSE) and can be used to gauge the relative difference between the same class of radicals, but also a good approximation of BDE, when added to the referent BDE. In Scheme 3, different relevant N-centered and C-centered radicals present in lactams and similar compounds (amidyl, aminyl, and alkyl radicals) are shown. A clear effect of sterical influence on the stability of amidyl radicals can be observed. Namely, for N-methyl-acetamide amidyl radical, RSE is 1.6 kJ mol–1, but when constricted in 2-azetidinone, 2-pyrrolidinone, 2-pepridinone, 2-azepanone, and 2-azocanone less stable amidyl radicals are formed, with RSEs in the range of 10.1 to 16.1 kJ mol–1. But as the ring grows, the structural effect on the radical is less pronounced, and for laurolactam amidyl radical (1-Nrad), RSE od –1.9 kJ mol–1 is between radical stability of N-methyl and N-ethyl (RSE = –6.6 kJ mol–1) derivatives of acetamide.
C-centered radicals are generally more stable than N-centered amidyl radicals (see Schemes 3 and 4). Alkyl radicals have RSE around that of C2-radical in propane (–27.6 kJ mol–1), ranging from –23.3 kJ mol–1 in C7-radical of 2-azocanone to –40.3 kJ mol–1 in C5-radical of 2-azepanone. While alkyl radicals surrounded by methylene neighbourghs are in the aforementioned range, C-centered radicals with carbonyl or amine part of amide next to it are more stable. The exceptions to that rule are radicals in β-lactam (2-azetidinone), which are heavily constrained and are among the least stable C-centered radicals, comparable only to the primary radicals.
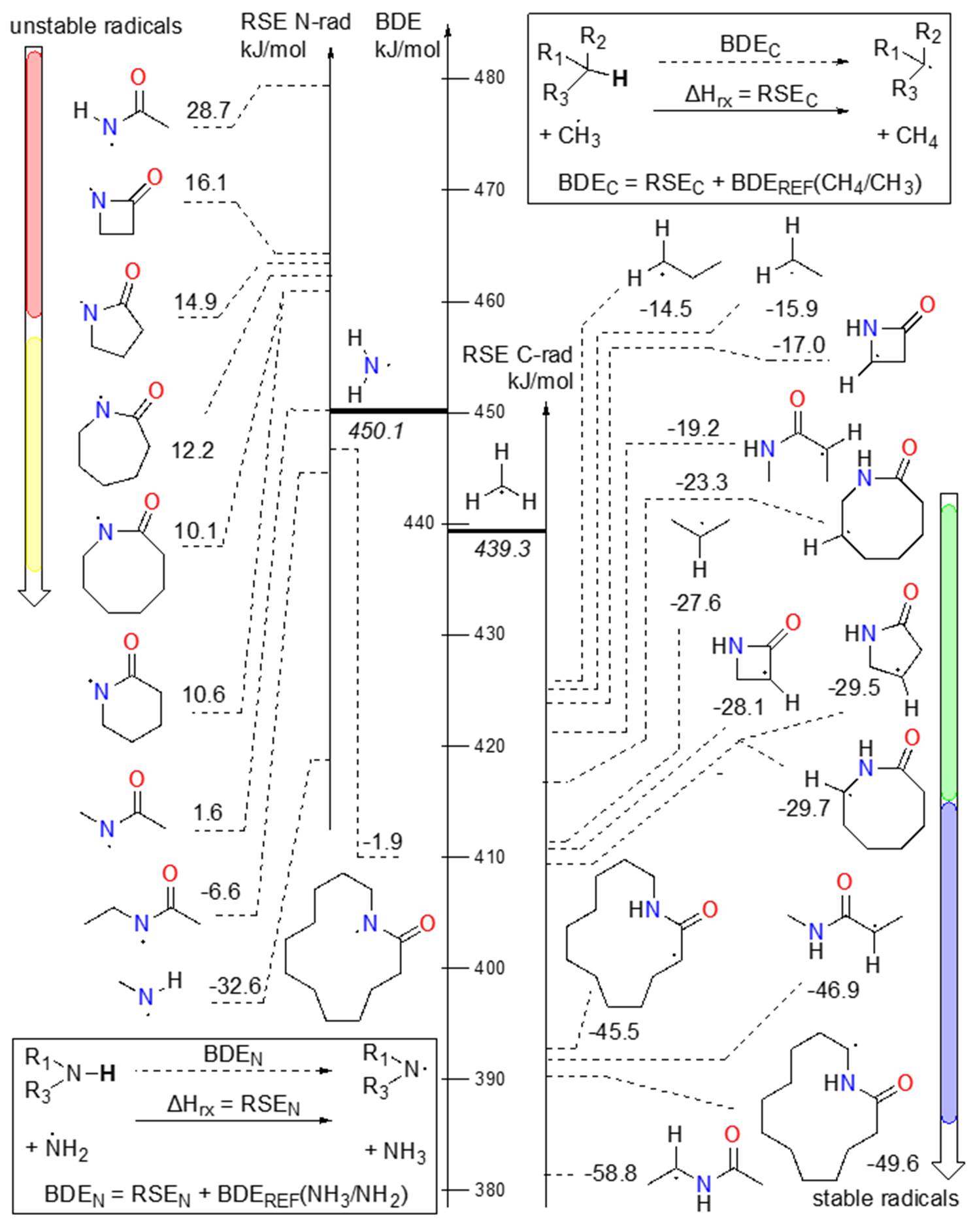
Scheme 3. BDE/RSE of selected N-centered radicals and C-centered radicals.
From Schemes 3 and 4 it is obvious that the HLF reaction is exothermic in the radical rearrangement reaction, and can occur in lactam rings of various sizes.
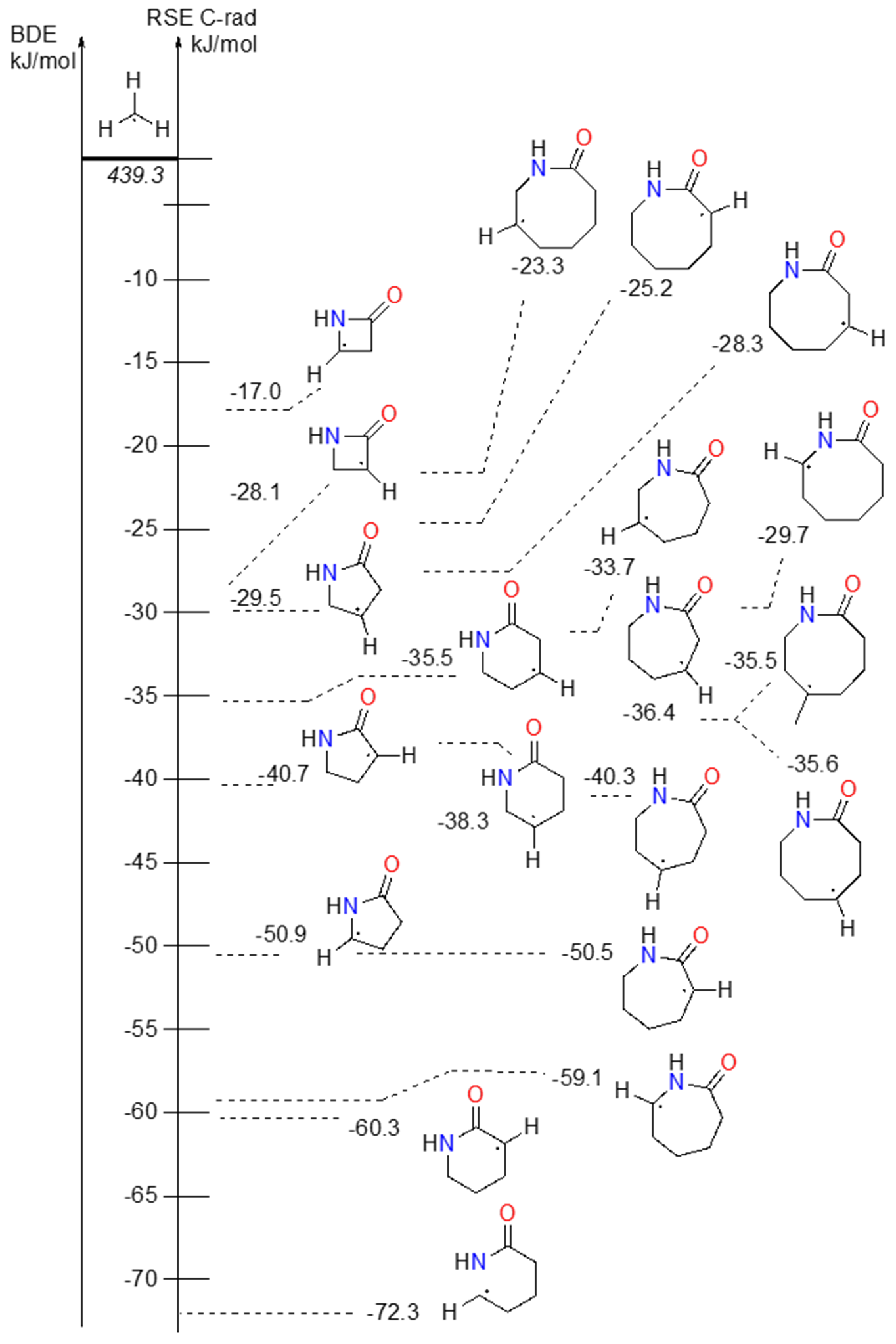
Scheme 4. BDE/RSE of C-centered radicals in lactam structures.
Next, we wanted to test our theory on a real macrocyclic lactam, specifically on laurolactam, in order to model all transition states and products stemming from the HLF reaction. The first step of the HLF reaction is light-induced homolytic cleavage of the N-halogen bond. To that end, an N-chloro derivative (1-Cl) of laurolactam (1) was prepared by N-chlorination with TCICA (Scheme 5).
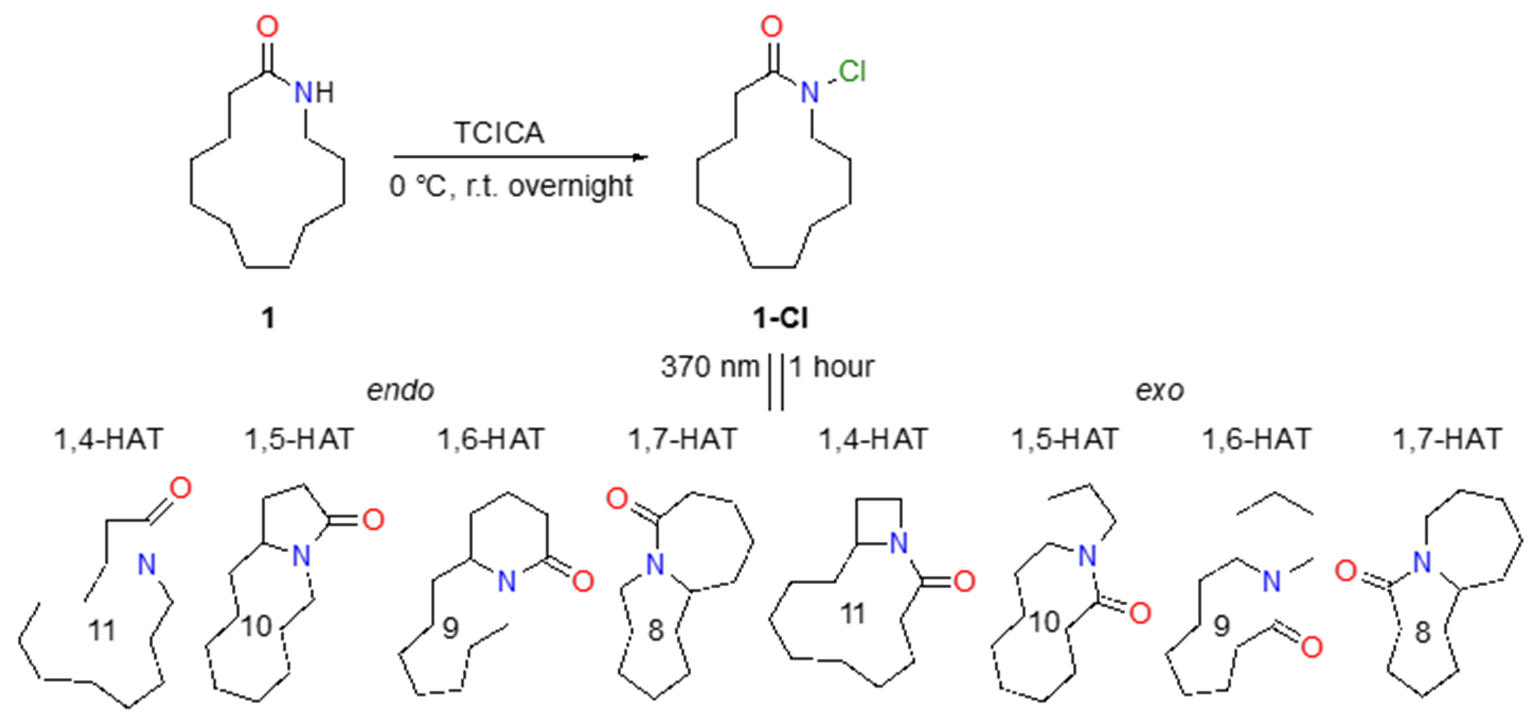
Scheme 5. N-chlorination of laurolactam and tentative products after 1-hour irradiation with 370 nm lamp.
Quantum-chemical methods were used to model all transition states and all products stemming from the HLF reaction. Possible 1,n-HAT reactions include 1,4-HAT, 1,5-HAT, 1,6-HAT, and 1,7-HAT forming 4-, 5-, 6-, and 7-membered rings, respectively (see Scheme 5). However, there are two different ways to form those rings. One pathway generates the product with the carbonyl group inside the smaller, newly formed ring, titled endo. Consequently, newly formed rings with the carbonyl group outside the smaller ring are termed exo. In Table I, calculated parameters are presented for all combinations. The lowest barriers (ΔH‡298) are present in the exo-1,5-HAT and exo-1,6-HAT for the cyclisation. The endo-1,5-HAT process is more stable than both endo-1,6-HAT and endo-1,7-HAT for endo-type of cyclisation, and very similarly stable to the 1,7-HAT path of exo-type. To our surprise, the barrier for the endo-1,4-HAT reaction is lower than the exo-1,4-HAT reaction. The formation of the β-lactam ring by 1,4-HAT endo-type of cyclisation is two orders of magnitude faster than the formation of azetidine by the exo-1,4-HAT, as seen from the calculated rate constants for endo- (kendo-1,4-HAT = 6.899 × 104 s–1) and exo- (kexo-1,4-HAT = 1.069 × 102 s–1) pathways of 4-membered ring cyclisations (transition-state structures are shown in Fig. 1). The stabilities of all products are similar and provide a strong thermodynamic force (reaction enthalpy, ΔHrx,298) towards products.
Table I. Calculated reaction parameters in the 1,n-HAT rearrangement reactions between amidyl N-centered and C-centered radicalsa
a The bottom row describes the formed ring in the subsequent reaction with the base.
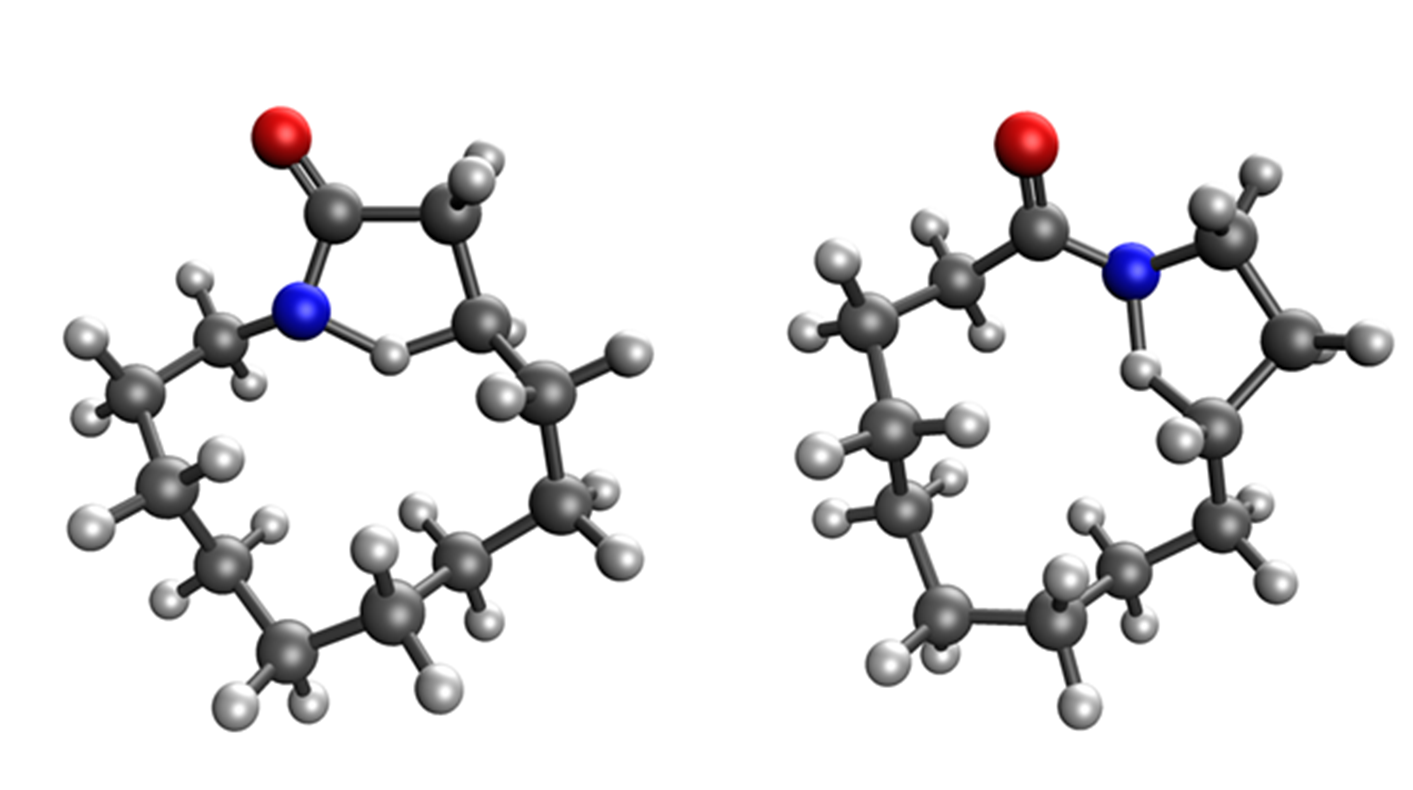
Fig. 1. Transition-state structures for endo-1,4-HAT and exo-1,4-HAT
Next, EPR experiments were performed as described in the method section. After spectra deconvolution, only two different radicals are observed. The major observed radical is the adduct of chlorine radical onto the N-tert-butyl-α-phenylnitrone (PBN), Cl-PBN, with hyperfine splitting constant (hfc) AN = 12.36 G, AH = 0.75 G and AH = 6.22 G, with a line width of 0.22. Another radical is a C-centered radical adduct of PBN, C-PBN, with a difference in g-value of –0.0013 from Cl-PBN, and hfc AN = 13.44 G, AH = 1.76 G. After decomposition, some residual signals remain with some weak and broad components which are similar to the N-PBN adduct radical observed in our previous work (15). Yet the residue signal is extremely weak, making it difficult to achieve a satisfactory deconvolution. Since the reactions happen very quickly, the rearrangement of N-centered radical to the C-centered radicals occurs faster than the spin-trapping reaction of PBN.
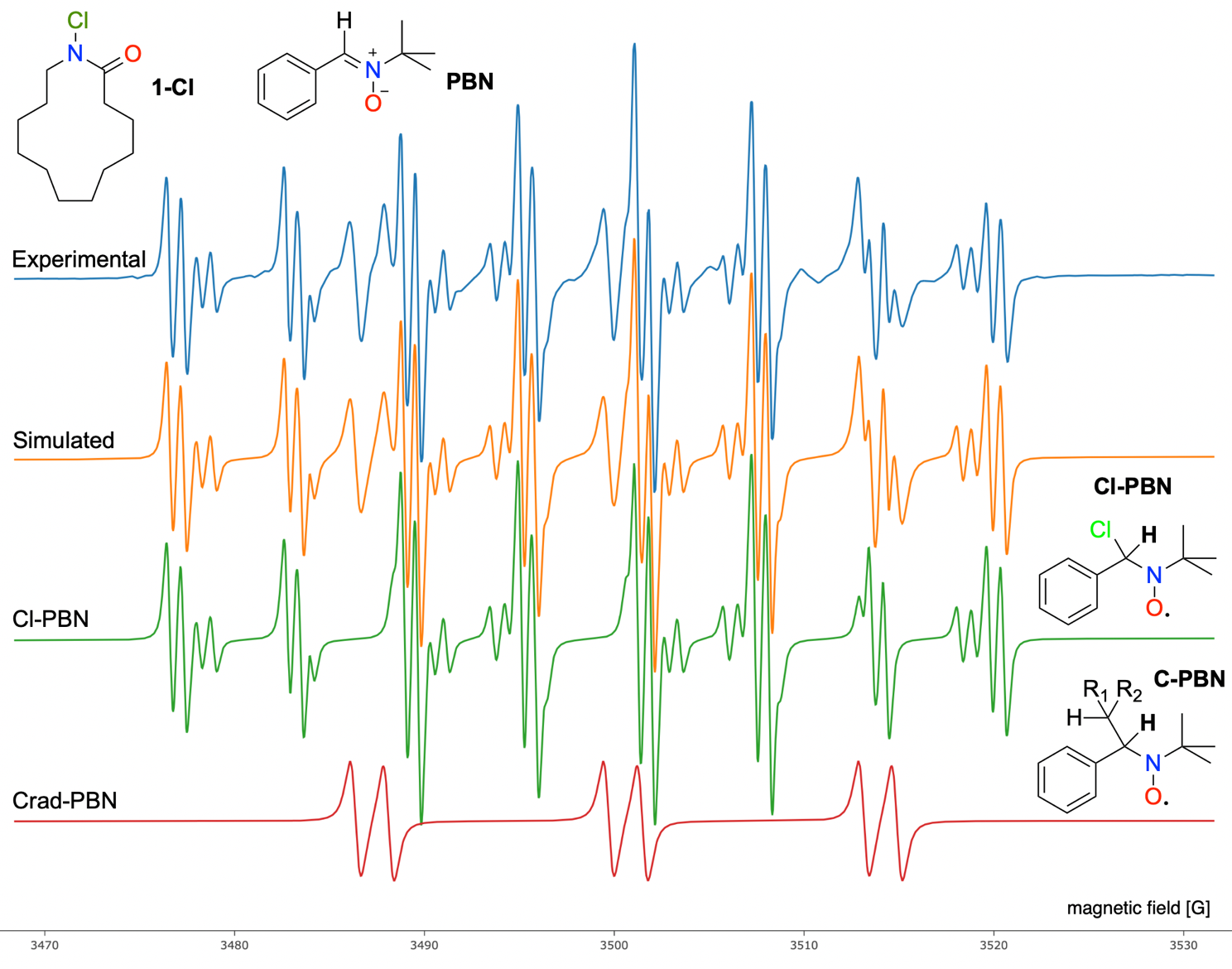
Fig. 2. EPR spectra of spin-trapped radical intermediates generated with a 370 nm irradiation of 1-Cl. The experimental spectrum is in blue, while red, green, and orange correspond to simulated spectra for Crad-PBN, Cl-PBN, and total simulated spectra, respectively.
Finally, NMR experiments were performed to identify the reaction products. The biggest obstacle encountered in the NMR experiments was the relative insolubility of the compound 1-Cl and its products in CDCl3 solvent. As the reaction progressed, precipitation was formed in the NMR tube, making prolonged spectra acquisition unfeasible. To address this issue, the photoreaction was quenched with 0.1 mol L–1 NaOH (added in excess), washed with water, and then 30 mg of the product mixture was resuspended in DMSO-d6 for NMR spectroscopy. In Fig. 3, APT-13C NMR spectra are shown, along with an enlarged portion of the spectra displaying signals of carbon atoms with an odd number of attached H-atoms. In the spectra, nine such signals can be observed, with a dubious signal at 62.05 ppm. If we disregard that signal, eight different signals actually correspond to the eight different products predicted via calculations (see Scheme 5).
Thus, we have demonstrated that HLF reaction can be used in the LSF strategies to produce all types of bicyclic rings from lactam macrocycles, with ring sizes ranging from 4-membered rings, up to 7-membered rings.
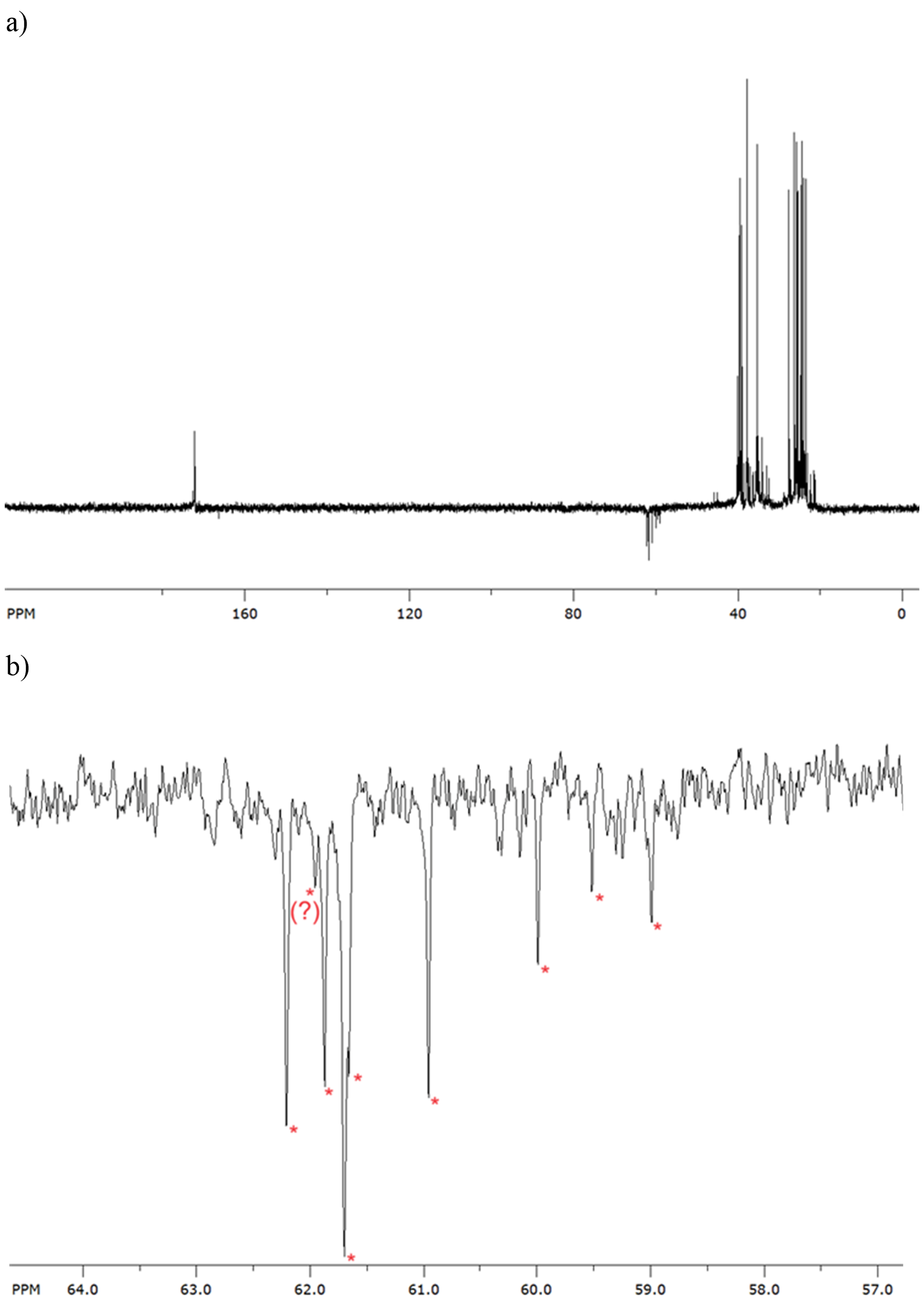
Fig. 3. a) APT-13C NMR spectra of the quenched and washed reaction; b) eight different signals in the negative region from 57–64 ppm correspond to eight different products.
CONCLUSIONS
This study successfully demonstrates the potential of the Hofmann-Löffler-Freytag (HLF) reaction as a viable method for late-stage functionalization (LSF) of macrocyclic lactams, as shown with laurolactam. The thermodynamically driven HLF reaction, characterised by radical stability differences, effectively transforms laurolactam into various bicyclic lactams with ring sizes ranging from 4- to 7-membered rings. These transformations, investigated with theoretical and experimental approaches, underscore the versatility and efficiency of the HLF reaction in generating structurally diverse and stable products. Amidyl radicals in constrained lactam rings exhibit higher radical stabilization energy (RSE) values, indicating reduced stability. C-centered radicals, particularly those adjacent to carbonyl or amine groups, demonstrate enhanced radical stability, with notable exceptions in highly constrained structures like 2-azetidinone. Computational modelling reveals the lowest activation barriers for 1,5- and 1,6-HAT exo-type cyclizations with carbonyl moiety outside the smaller ring. The unexpected lower barrier for 1,4-HAT endo-type reactions compared to exo-type counterparts highlights the complexity and potential of the HLF mechanism in the formation of pharmacologically potent β-lactam motif. EPR spectroscopy identifies Cl-PBN and C-PBN as primary radical adducts to the spin-trap N-tert-butyl-α-phenylnitrone (PBN), underpinning rapid N- to C-centered radical rearrangement. NMR spectroscopy confirms the formation of eight distinct products, aligning with computational predictions. The findings affirm the HLF reaction's applicability in LSF strategies, enabling the production of various bicyclic rings from macrocyclic lactams. This approach not only broadens the scope of accessible ring structures but also facilitates the fine-tuning of pharmacological properties, crucial for drug development. Future research should focus on exploring different macrocyclic substrates, optimising reaction conditions, and conducting biological evaluations to fully harness the HLF reaction's potential in drug development.
Abbreviations, acronyms, symbols. – APT, attached proton test; BDE, bond dissociation energy; EPR, electron paramagnetic resonance; ΔH‡298, activation enthalpy at 298K; ΔHrx,298, reaction enthalpy at 298K; HAT, hydrogen atom transfer; hfc, hyperfine coupling constant; HLF, Hofmann-Löffler-Freytag; LED UV, light-emitting diode in ultraviolet radiation; L SF, late-stage functionalization; NMR, nuclear magnetic resonance; PBN, N-tert-butyl-α-phenylnitrone; RSE, radical stabilization energy; TCICA, trichloroisocyanuric acid.
Acknowledgments. – The authors would like to acknowledge financial support from the Croatian Science Foundation Installation grant UIP-2020-02-4857 LIGHT-N-RING, grant IP-2022-10-2634 PharmaEco, and computational resources available on Cluster Supek, EU funded through KK.01.1.1.08.0001, a part of the Advanced Computing Service provided by University of Zagreb University Computing Centre – SRCE. This work was also supported by the project FarmInova at the Faculty of Pharmacy and Biochemistry University of Zagreb (KK.01.1.1.02.0021) funded by the European Regional Development Fund. Support from The Cryogenic Centre at the Institute of Physics project (KaCIF, KK.01.1.1.02.0012) is acknowledged.
Funding. – This research received no external funding.
Conflicts of interest. – The authors declare no conflict of interest.
Authors contributions. – Conceptualization, D.Š. and G.Z.; calculations, data analysis, G.Z.; methodology, experimental, J.Y., K.P. and T.P; analysis, T.P. and V.V.; writing, original draft preparation, D.Š.; writing, review and editing, E.B. and D.Š.; funding, D.Š. All authors have read and agreed to the published version of the manuscript.