
INTRODUCTION
The increasing focus on free radical species in the life sciences, combined with the unique capability of electron paramagnetic resonance (EPR) spectroscopy to provide direct evidence of free radicals, has spurred heightened interest in this spectroscopic method across various medical and biomedical disciplines. Free radicals, particularly reactive oxygen species such as hydroxyl radicals (•OH), superoxide anions (O2•−), alkoxyl radicals (R-O•), and peroxyl radicals (R-OO•), have been identified as crucial signaling molecules (1, 2). When these radicals exceed the capacity of antioxidant defenses, they can act as pathogenic agents contributing to a range of diseases, including inflammatory responses and ischemia/reperfusion injury (3–6). Consequently, EPR spectroscopy has become an invaluable tool in the pharmaceutical sector, particularly in drug discovery, for investigating natural and synthetic compounds with radical scavenging properties, which can serve as potential preservatives to extend the shelf life of pharmaceuticals (7, 8). Furthermore, EPR spectroscopy has proven essential in elucidating the mechanisms of action of numerous drugs that operate through radical intermediates, such as anticancer, antimalarial, or NO-releasing drugs (9–12).
Additionally, EPR spectroscopy represents an indispensable tool in pharmaceutical development, particularly due to its capacity to elucidate a range of critical processes. Its applications encompass the study of drug-membrane interactions and oxidative processes, both in vitro and in vivo (13, 14), as well as the analysis of drug metabolism and pharmacokinetics (15). EPR spectroscopy is instrumental in characterizing drug delivery systems (16–18), detecting microacidity (19, 20), and examining phase transitions (21, 22).
In the realm of pharmaceutical science, EPR spectroscopy is crucial for addressing issues related to single-electron chemistry and impurity tracking (23–25). Its efficacy extends to the examination of paramagnetic anticancer metallodrugs, offering valuable insights into their mechanisms of action, conformational dynamics, ligand exchange processes, pH effects, and redox reactions (26–29). Furthermore, EPR’s versatility supports quantitative analysis, enabling drug detection in body fluids, pharmacokinetic studies, and tissue oximetry (30, 31). As EPR spectroscopy continues to advance, its role in all the aforementioned areas of pharmaceutical science is anticipated to become increasingly significant.
This review provides an overview of the diverse applications of EPR spectroscopy within the pharmaceutical field. EPR spectroscopy can significantly contribute to the advancement of pharmaceutical research and help ensure the integrity and efficacy of drug products. With a selection of representative examples, the utility and versatility of EPR spectroscopy in specific phases of drug discovery, development, and production, as well as in post-market quality control studies will be demonstrated.
BASIC PRINCIPLES OF EPR SPECTROSCOPY
EPR is a spectroscopic technique that specifically detects species containing unpaired electrons by monitoring the absorption of microwaves when these paramagnetic species are placed in an external magnetic field. Measured species include free radicals (e.g., superoxide radicals), odd electron molecules (e.g., nitric oxide), transition-metal complexes (e.g., cis-platin), and lanthanide ions (e.g., gadolinium as a contrasting agent in CT).
The fundamental principles of EPR spectroscopy closely resemble those of the more widely known nuclear magnetic resonance (NMR) spectroscopy. Both techniques rely on the interaction of electromagnetic radiation with magnetic moments, which are generated by electrons in EPR spectroscopy and by nuclei in NMR spectroscopy. In the case of EPR spectroscopy, the magnetic moment of an unpaired electron originates from its intrinsic spin, stemming from its quantum mechanical properties. When subjected to an external magnetic field, the electron spin aligns either parallel or antiparallel to the direction of the field, corresponding to a lower ( –½) or higher ( +½) energy state, respectively.
The energy difference () between these two spin states is directly proportional to the strength of the applied magnetic field () and can be expressed by the equation:
(1)
where is Planck's constant, is the frequency of the electromagnetic radiation, is a dimensionless constant known as the g-factor (with a value of 2.0023 for an unpaired electron and slightly modified for electrons in molecules), and is the Bohr magneton.
The orbital and spin motion of an electron gives rise to an additional magnetic field, which exerts an influence on the unpaired electron. This supplementary magnetic field is denoted as . Incorporating this additional field into the Equ. 1, we obtain:
(2)
where represents the g-factor for a free electron in a vacuum, while describes the effects of the environment on the electron (Eq. 2). The modified g-factor, , serves as a unique identifier, incorporating the chemical information about the interaction between the electron and its surrounding chemical environment.
In other words, the g-factor is a dimensionless parameter that characterizes the magnetic moment and angular momentum of an unpaired electron in a paramagnetic substance. It delineates the splitting of an electron's energy levels under the influence of an external magnetic field, but more importantly, it provides critical insight into the electronic environment of the unpaired electron and can be used for the identification of particular radical species. Experimentally, the g-factor is determined by measuring the resonance condition (Eq. 1) in an EPR experiment.
When electromagnetic radiation with an energy corresponding to the difference described in Eq. 1 is applied to a sample, resonance transitions between the lower and upper energy states become feasible. This process involves the absorption of microwave radiation, generally within the 9–10 GHz range, which provides the energy necessary for an unpaired electron to transition between its two magnetic energy levels. Fig. 1 illustrates the Zeeman splitting of spin energy levels when a paramagnetic system is placed in an external magnetic field.
The resonance frequency in EPR spectroscopy is approximately 103 times higher than in NMR spectroscopy due to the larger magnetic moment of the electron, which accounts for the greater sensitivity of EPR spectroscopy in comparison to NMR spectroscopy. EPR spectroscopy can detect free radical concentrations as low as 0.5–1.0 nmol mL–1, whereas NMR spectroscopy typically requires much higher concentrations due to the weaker magnetic moments of nuclei.
The higher frequency and greater sensitivity of EPR spectroscopy make it especially valuable for studying paramagnetic species, including free radicals, transition metal complexes, and defects in solids (32, 33), where the larger magnetic moment of electrons provides more detailed insights into the electronic environment of the sample.
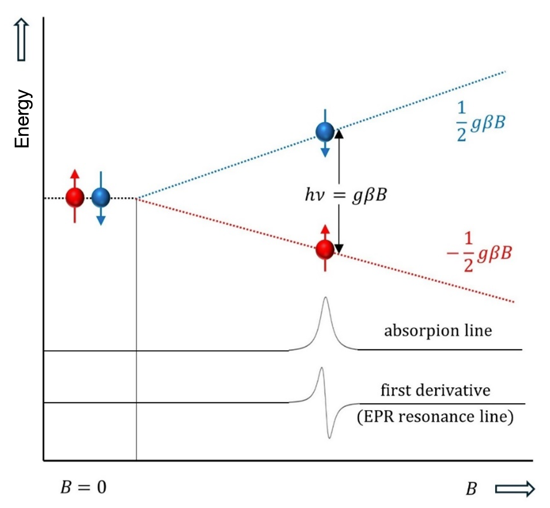
Fig. 1. Basic principle of EPR spectroscopy—absorption of microwave radiation.
In addition to the g-factor, hyperfine interaction represents an important parameter in EPR spectroscopy. Atomic nuclei within a molecule possess magnetic moments, which can generate a local magnetic field sufficiently strong to influence the electron. The interaction between an unpaired electron and adjacent magnetic nuclei leads to splittings in the EPR spectrum, known as hyperfine splittings. Hyperfine interaction is instrumental in elucidating the number and identity of atoms within a molecule or compound, as well as their spatial relationship to the unpaired electron. The quantity and nature of the interacting nuclei determine the number of spectral lines and their respective intensities. For instance, the magnetic interaction between the unpaired electron and the nuclear spin of nitrogen (, ¹⁴N) in nitroxyl radicals leads to hyperfine splitting into three distinct lines, termed triplets. Conversely, when the unpaired electron interacts with a proton, the EPR spectrum displays only two lines, or doublets, reflecting the interaction with the proton’s different nuclear spin ( ½). Generally, hyperfine interaction between an unpaired electron and nuclei with a non-zero spin causes the primary resonance line to split into lines, where represents the number of nuclei in the electron's vicinity. The g-factor and hyperfine splitting are fundamental EPR parameters used to characterize the structure of the paramagnetic species under investigation.
In the context of nitroxyl radicals, which are frequently studied in biological systems using EPR spectroscopy, the unpaired electron is not entirely localized within the –orbital of the nitrogen atom. Instead, it may also be localized on the oxygen atom or delocalized across the ring structure of the nitroxyl radicals. This delocalization renders nitroxyl radicals highly sensitive to subtle charge density variations induced by surrounding molecules. The extent of the interaction between the electron and the nuclei, along with their magnetic moments, determines the distance between the hyperfine splitting lines. This separation between peaks, denoted by the hyperfine splitting constant (), is a critical descriptor in EPR analysis. Additionally, other easily obtainable parameters from EPR spectra include the signal amplitude of the peaks () and the peak-to-peak line width (). Fig. 2 illustrates the simulated EPR spectrum of the nitrogen spin probe, in which three important EPR parameters are indicated: hyperfine splitting constant, signal amplitude, and peak-to-peak line width.
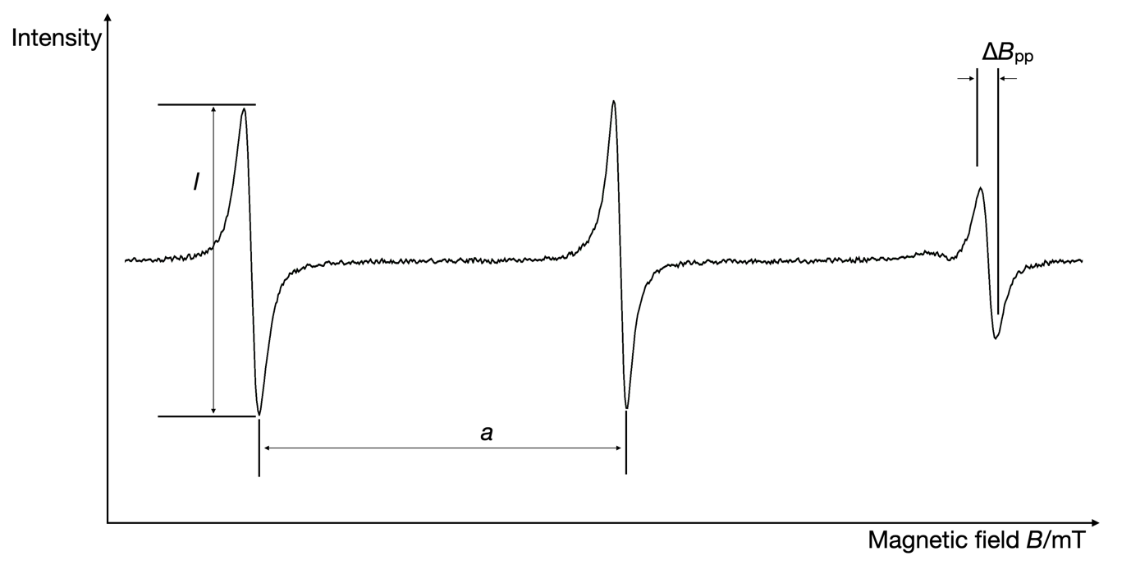
Fig. 2. Simulated EPR spectrum of the nitrogen spin probe illustrating typical EPR parameters: hyperfine splitting constant; —signal amplitude; —peak-to-peak line width.
EPR spectra are typically obtained as first-derivative signals. Consequently, the intensity of the signal is determined through the process of double integration of the EPR spectra; the first integration provide classical absorption spectra, whereas the second integration of the peaks provide the quantified intensity of the signal.
SPIN TRAPPING TECHNIQUE
Spin trapping is an essential technique in EPR spectroscopy, used to detect and characterize short-lived free radicals that are typically challenging to observe directly. The technique involves employing a "spin trap", a stable EPR silent molecule that selectively reacts with transient radicals, forming a more stable spin adduct with an EPR signal. This adduct, often a nitroxide-type radical, is sufficiently stable to allow for subsequent detection and analysis through EPR spectroscopy.
The methodology of spin trapping comprises several key components: the selection of an appropriate spin trap, the generation of radicals, the formation of the spin adducts, detection using EPR, and finally analysis and interpretation of this adduct. Nitrones and nitroso compounds, such as 5,5-dimethyl-1-pyrroline N-oxide (DMPO) and N-tert-butyl-α-phenylnitrone (PBN), are commonly used spin traps. These compounds react rapidly with radicals generated through various processes, including chemical reactions, radiation, or enzymatic activity, to form stable spin adducts (34, 35). Database of commonly used spin-traps with detected spin-adducts are stored at the National Institute of Environmental Health Sciences (NIEHS database: https://tools.niehs.nih.gov/stdb/index.cfm). Fig. 3 shows the chemical structures of the two most used spin traps, DMPO and PBN.
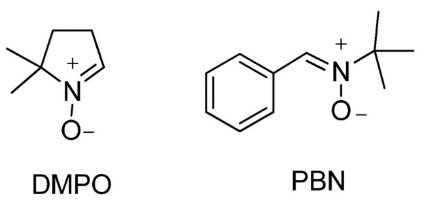
Fig. 3. The chemical structure of the commonly used spin traps 5,5-dimethyl-1-pyrroline N-oxide (DMPO) and N-tert-butyl-α-phenylnitrone (PBN).
The EPR spectral analysis of the spin adducts, particularly focusing on hyperfine splitting patterns and g-factors, enables the identification and characterization of the original radical species. For example, in a typical experiment, hydroxyl radicals (•OH) might be generated and subsequently trapped by DMPO, forming a DMPO-OH adduct (36, 37). The EPR spectrum of this adduct offers critical insights into the presence and nature of hydroxyl radicals. Notably, the spectrum exhibits a characteristic quartet, with two central lines possessing double the intensity of the two peripheral lines (1:2:2:1 quartet). Given that this is a nitrogen-centered radical, one would expect a triplet pattern, corresponding to the nitrogen nuclear spin (). However, partial interaction of the electron spin with the hydrogen nucleus ( ½) results in each line being further split into a doublet, theoretically yielding a sextet in the overall spectrum. Due to the hyperfine coupling constants of the nitrogen and hydrogen nuclei being equivalent ( mT), the resulting spectrum is an overlapped pattern, collapsing the expected six lines into a four-line spectrum with an intensity ratio of 1:2:2:1, as shown in Fig. 4a. An additional example is illustrated in Fig. 4b, where the 4-methylphenyl radical reacts with the DMPO spin-trap, forming an adduct with hyperfine couplings of 1.454 mT and 2.134 mT. In this case, the absence of overlapping lines produces the expected sextet pattern (38).
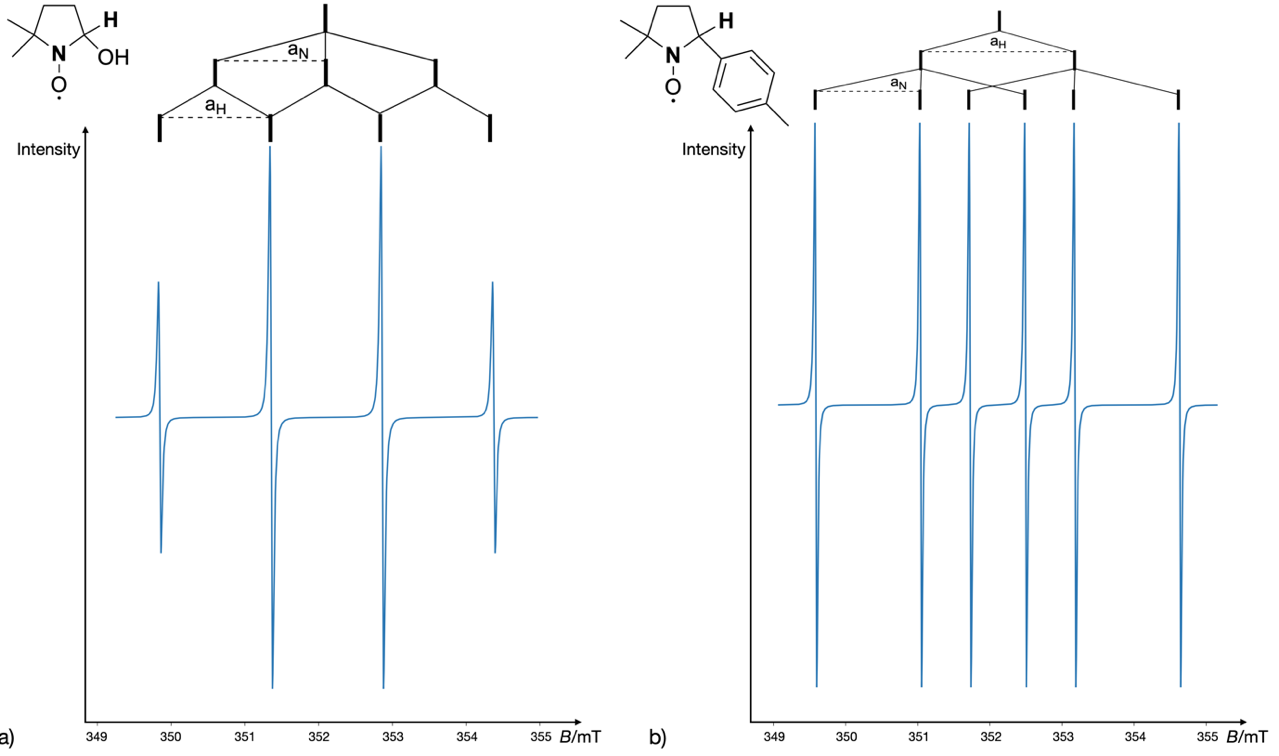
Fig. 4. The simulated EPR spectrum of: a) the DMPO-OH adduct, and b) the 4-methylphenyl radical spin adduct of DMPO. VisualEPR program was used to simulate the experimental data as reported in ref. 38.
Spin trapping is extensively applied across diverse scientific disciplines. In biological research, it is used to detect reactive oxygen species (ROS), while in chemistry, it facilitates the study of radical-mediated reaction mechanisms. As an example of the latter, Fig. 5 displays the EPR spectra of spin adducts of several radicals generated in the variant of the Hofmann-Löffler-Freytag (HLF) reaction, triggered by UV light-induced activation (39, 40). The HLF reaction is a potential late-stage functionalization strategy for the efficient conversion of macrocyclic to bicyclic rings in lactams, proposed as an additional approach to further enhance the metabolic stability of peptide-based drugs (unpublished result of our group).
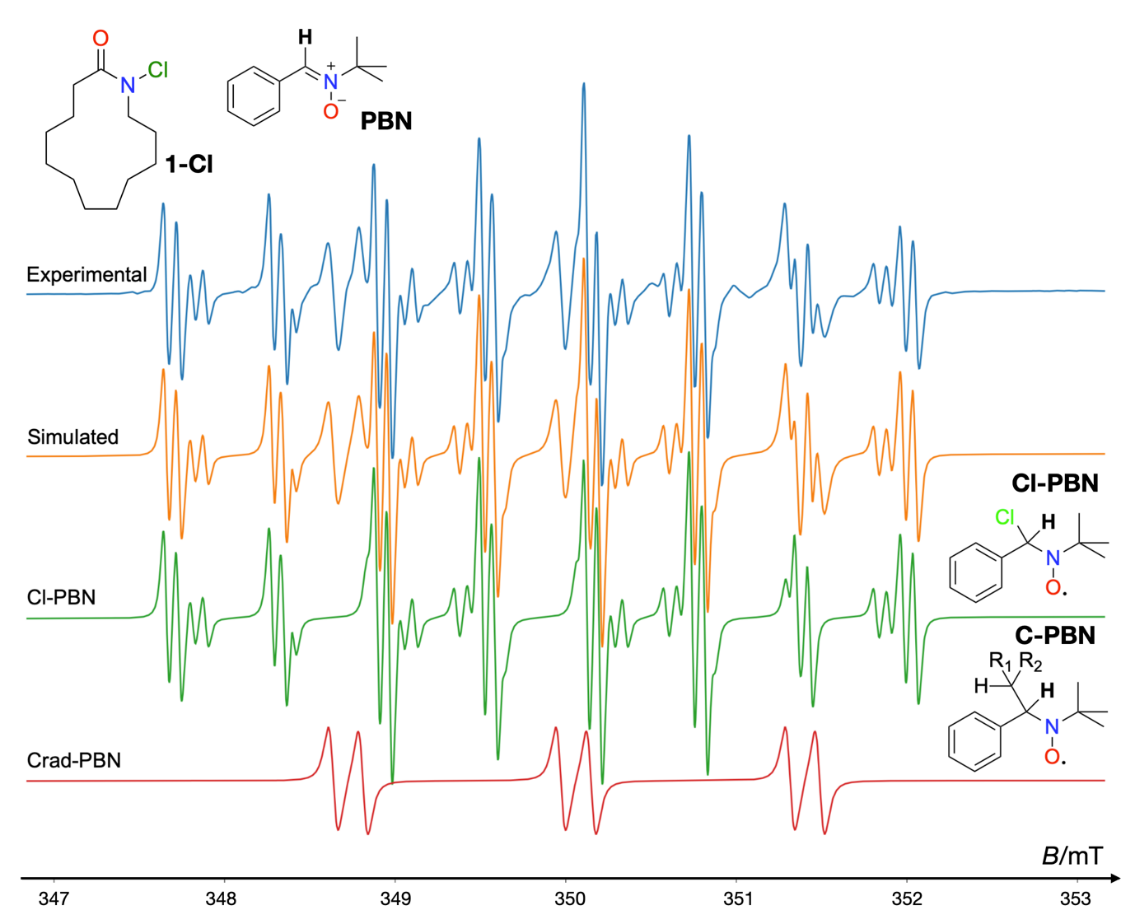
Fig. 5. EPR spectra of spin-trapped radical intermediates generated with 370 nm irradiation of 1-chloroazacyclotridecan-2-one (1-Cl). The experimental spectrum is in blue, while red, green, and orange correspond to simulated spectra for the C-centered radical adduct of PBN (Crad-PBN), Cl-centered radical adduct of PBN (Cl-PBN), and total simulated spectra, respectively.
Overall, by capturing otherwise elusive and short-lived radicals through the formation of stable spin adducts, this methodology enables the analysis of species that would otherwise be unattainable. Consequently, the spin trapping technique offers profound insights into the identity, concentration, and formation mechanisms of transient radicals, highlighting its significance in the study of radical chemistry and related fields.
SPIN LABELING (SPIN PROBE) TECHNIQUE
Spin labeling and spin-probe techniques are used in EPR spectroscopy to study the structure, dynamics, and interactions of biomolecules or other complex systems (41, 42). It involves the attachment of a stable free radical, referred to as a "spin label", or the introduction of a stable free radical, known as a "spin probe", to a specific site on the molecule of interest. Spin labels are stable, paramagnetic molecules, often free radicals or metal ion complexes, that are covalently attached to a specific site within a molecule (such as proteins, lipids, or DNA). Nitroxyl radicals are again commonly used as spin labels due to their unpaired electron, which makes them detectable by EPR spectroscopy. In contrast to spin labels, spin probes are small paramagnetic molecules that are not covalently attached to any particular site but are instead introduced freely into the system. These probes are used to monitor the overall properties of the environment or medium in which they are introduced. They provide information about the general characteristics of the system, such as solvent polarity, viscosity, temperature, or the molecular environment in biological membranes. Unlike spin labels, which are localized to a specific site, spin probes offer a broader assessment of the properties of the surrounding medium. They are commonly employed in EPR spectroscopy to study the behavior of molecular systems in a non-specific manner, providing valuable insights into the physical properties of the medium rather than localized interactions. The most used spin probe is 2,2,6,6-tetramethylpiperidine-1-oxyl (TEMPO), the chemical structure of which is depicted in Fig. 6. The major distinct point between spin traps and spin labels or spin probes is that the spin traps are EPR silent until radical reacts with them, while spin labels or spin probes are EPR active (have EPR spectra) as soon as they are dissolved.
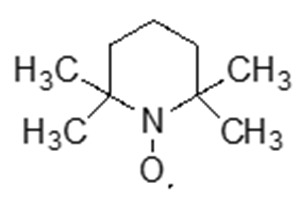
Fig. 6. The chemical structure of 2,2,6,6-tetramethylpiperidine-1-oxyl (TEMPO), the most used spin probe in EPR spectroscopy.
Once the spin label is attached or the spin probe is introduced to the system of interest, EPR spectroscopy can be used to monitor the behavior of the labeled molecule. The resulting EPR spectra provide insights into the local environment of the spin label or the spin probe, including information on molecular motion, microviscosity, and the polarity of the surroundings (43, 44). By analyzing these spectra, researchers can infer structural details, conformational changes, and dynamic processes occurring within the labeled molecule or its environment. This makes spin labeling a valuable tool in fields such as biochemistry, structural biology, and material science. Besides polarity, binding interaction, temperature, and molecular size, the tumbling behavior of spin probes is highly dependent on the microviscosity of their surroundings (44, 45). In low-viscosity media, spin probes exhibit rapid free tumbling, resulting in highly symmetric EPR spectra characterized by narrow lines and short rotational correlation times 𝜏c. As the viscosity increases, the tumbling rate decreases, leading to restricted motion, line broadening, and increased rotational correlation times values. Representative simulated EPR spectra of a spin probe in environments with varying microviscosities are shown in Fig. 7. These changes in the EPR spectra of spin probes offer a direct method for assessing the viscosity and other dynamic properties of the environment (46).
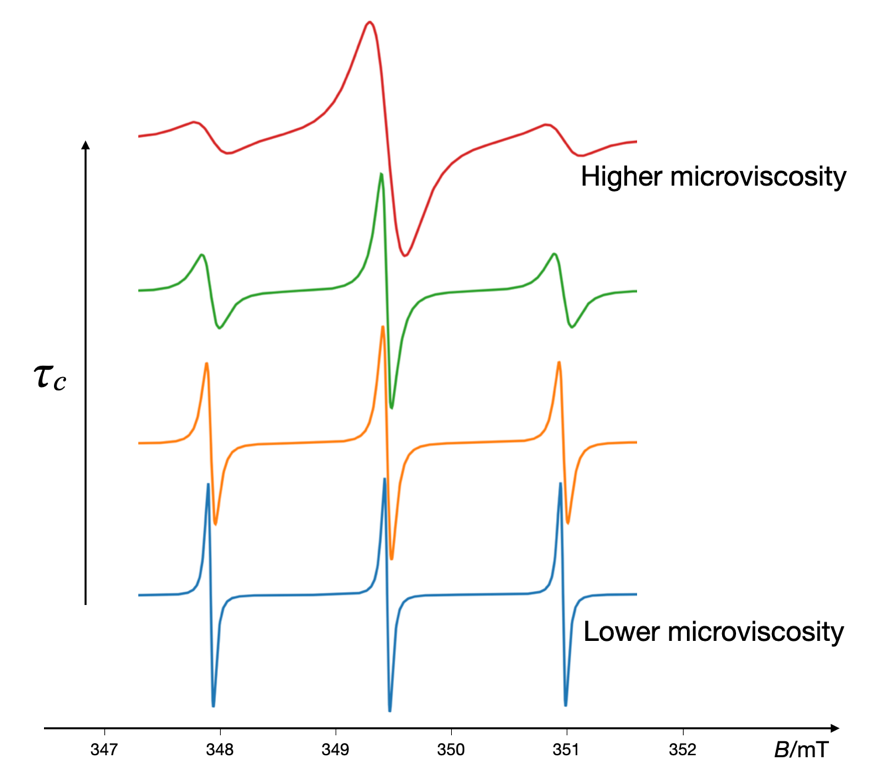
Fig. 7. Representative simulated EPR spectra of the nitroxyl radical in environments with varying microviscosities. The microviscosity of the spin probe's surroundings is inferred from the corresponding rotational correlation times (𝜏c), where increased correlation times signify reduced tumbling rates, indicative of higher microviscosities. The VisualEPR program was employed to simulate the EPR spectra of the spin probe in various environments.
In summary, spin labels and spin probes, through their EPR spectra, offer a powerful tool for probing the microviscosity, polarity, and molecular dynamics of their environments. The principles of molecular tumbling and hyperfine interactions are directly applicable to understanding and interpreting the behavior of spin labels in various contexts, making them indispensable in the study of complex systems.
EPR ANALYSIS OF FREE RADICAL SCAVENGING PROPERTIES IN PHARMACEUTICAL COMPOUNDS
Free radicals, characterized by unpaired electrons, play a pivotal role in various physiological and pathological processes, including oxidative stress, inflammation, and cellular damage (47–49). Their contribution to oxidative damage of macromolecules such as DNA, proteins, and lipids is strongly linked to the development of numerous degenerative diseases, including cancer, atherosclerosis, neurodegenerative disorders, and diabetes (50, 51). ROS and reactive nitrogen species (RNS), which are naturally produced during cellular metabolism, further exacerbate oxidative stress, initiating harmful chain reactions. Consequently, the scavenging of these radicals has become a crucial strategy in pharmaceutical development, particularly in the design of antioxidant compounds that aim to mitigate oxidative damage and restore cellular homeostasis (52).
EPR spectroscopy has emerged as an indispensable tool for investigating the radical scavenging potential of both pharmaceutical and natural antioxidant compounds. It is especially effective in detecting key radicals like hydroxyl (•OH) and superoxide (O2•−), which play central roles in oxidative damage (53–56). By exposing these radicals to drugs or antioxidants and measuring the resultant reduction in radical concentration, EPR spectroscopy provides precise, real-time insights into the antioxidative efficacy of these compounds. The EPR signal intensity is directly proportional to the concentration of free radicals, making it a reliable method for quantifying antioxidant properties (57). This approach involves measuring the variations in EPR signal intensity of stable radicals following their interaction with antioxidant compounds (58). Several well-known radicals, including 1,1-diphenyl-2-picrylhydrazyl (DPPH), 4-hydroxy-2,2,6,6-tetramethyl-1-piperidinyloxy (TEMPOL), and galvinoxyl, have been utilized in such studies (59, 60). The chemical structures of these radicals, as well as the EPR spectra of DPPH and TEMPOL, are well-documented, providing a framework for assessing antioxidant interactions. Fig. 8 illustrates the chemical structure of stable radicals commonly employed for the measurement of antioxidant properties via EPR spectroscopy. Fig. 9 presents simulated EPR spectra of a typical DPPH scavenging experiment, demonstrating the time-dependent decay of the five-line EPR spectrum of DPPH.
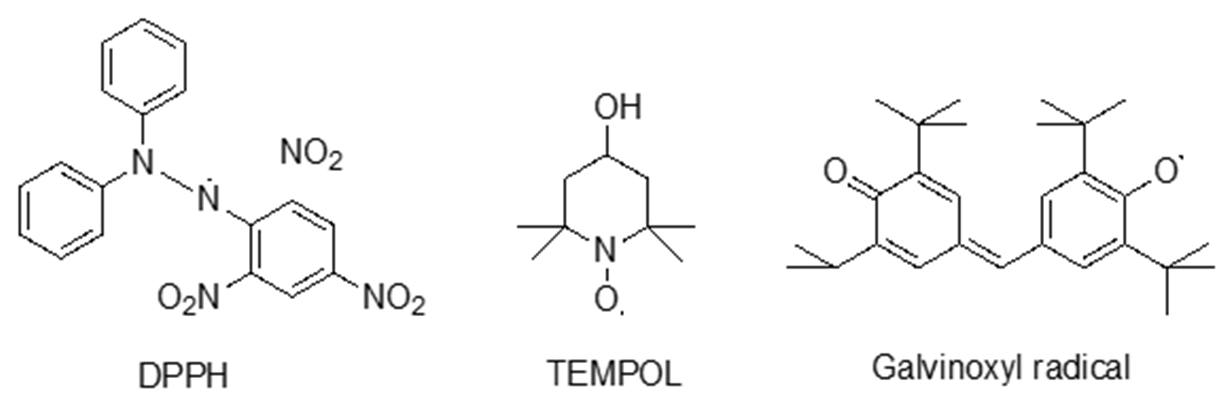
Fig. 8. Chemical structures of stable free radicals commonly used for the measurement of antioxidant properties by EPR spectroscopy.
The scavenging properties of pharmaceutical compounds can be evaluated by exposing known free radicals, such as superoxide (O2•−), hydroxyl (•OH), or nitric oxide (NO•), to the investigated drug (61, 62). EPR spectroscopy is then employed to quantify the reduction in radical concentration as a function of time or dose. The decay rate of the EPR signal correlates with the compound's scavenging potential, providing a direct measure of its antioxidant properties (55).
EPR spectroscopy can be used to monitor the disappearance of radical signals as a result of interaction with a pharmaceutical agent. Spin trapping techniques, as described above, further enhance the sensitivity of EPR spectroscopy in such studies (63). By tracking the decay of these EPR signals over time, the efficiency of a compound's scavenging properties can be quantified, while changes in the spectral profile may offer insight into specific reaction mechanisms (64).
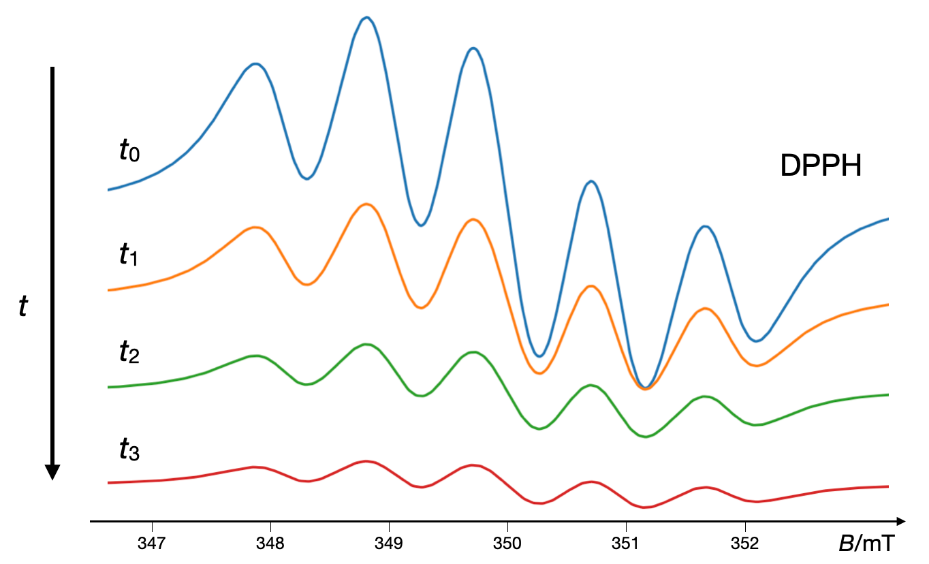
Fig. 9. A typical DPPH scavenging EPR experiment showing the decay of the simulated five-line EPR spectrum of DPPH over time. The simulation was performed using the VisualEPR program.
Certain pharmaceutical compounds are renowned for their antioxidant properties, acting through mechanisms such as direct free radical scavenging, metal chelation, or the maintenance of endogenous antioxidant systems (65, 66). Various analytical techniques, including DPPH, NBT, and ABTS assays, traditionally assess antioxidant properties by tracking changes in absorbance as radicals are reduced (67, 68). However, EPR spectroscopy surpasses these methods in specificity and versatility, and offers several distinct advantages over traditional methods for assessing antioxidant properties. First, EPR spectroscopy enables the direct and highly specific detection of free radicals, providing superior specificity compared to alternative techniques. Second, unlike colorimetric methods, EPR spectroscopy is unaffected by the presence of pigments, opacity, or suspended particles, ensuring accurate measurements even in complex extracts. Third, EPR spectroscopy demonstrates remarkable versatility, particularly in measuring the scavenging properties of short-lived radicals via spin trapping techniques, which enhances the sensitivity of the analysis. Furthermore, modern EPR spectrometers can incorporate temperature modulation or UV irradiation, facilitating detailed thermal and photo-stability studies (69). Fourth, EPR spectroscopy is applicable to both solid and liquid samples, broadening its range of use. From the above-mentioned attributes, it can be concluded that EPR spectroscopy surpasses colorimetric methods in their suitability for analyzing antioxidant properties in concentrated samples and should be used more often in routine analysis. As newer benchtop EPR instruments become available, which offer satisfactory signal-to-noise ratios and sensitivity, EPR techniques can be employed on-site, without special requirements, such as continuous running water for the maintenance of constant cooling of the magnet and power supply. In certain instances, these instruments can even be integrated directly with the flow apparatus.
EPR spectroscopy has been widely applied to investigate the antioxidant properties of vitamins E and C, both of which play crucial roles in protecting biological systems from oxidative stress (70, 71). Vitamin E, a lipid-soluble antioxidant, has been studied for its ability to neutralize lipid peroxyl radicals in cell membranes, with EPR spectroscopy revealing how it regenerates from its oxidized form (α-tocopheroxyl radical) through interactions with other antioxidants, such as vitamin C (72). Vitamin C, a water-soluble antioxidant, has been extensively analyzed using EPR spectroscopy for its ability to directly scavenge reactive oxygen species, regenerate vitamin E, and protect against oxidative DNA damage (73). These studies have provided key insights into the synergistic effects of these vitamins and their roles in maintaining redox balance in various biological environments.
EPR spectroscopy has been previously utilized to evaluate the radical scavenging activity of catechins, tea components, wines, cognacs, fruits, and beer (55, 74–78). Moreover, it has been instrumental in investigating the oxidizing effectiveness of superoxide radicals and the stability of antioxidants in microemulsions (79). A recent review highlights the use of modern analytical methods, including EPR spectroscopy, in pharmaceutical research to identify potent antioxidants that protect human skin from UV radiation-induced damage (80).
The antioxidant mechanisms of stobadine, a pyridoindole derivative known for its cardioprotective and antihypoxic properties, have been investigated through EPR spectroscopy (81). Oxidation of stobadine by PbO₂/tBuOOH in benzene results in the formation of a nitroxide radical, detectable by EPR spectroscopy at room temperature. This observation signifies the conversion of the indolic amino group into the corresponding nitroxide radical. In another study, the quantitative free radical scavenging properties of curcumin and its demethoxy derivatives (demethoxycurcumin and bisdemethoxycurcumin) as well as its hydrogenated derivatives (tetrahydro-curcumin, hexahydro-curcumin, and octahydro-curcumin) against DPPH, NO• radical, hydroxyl radical, and superoxide anion radical, using EPR spectroscopy (82). The results indicated that curcumin and its derivatives primarily function as chain-breaking antioxidants rather than direct free radical scavengers. Additionally, the ortho-methoxyphenolic group and heptadione linkage in these compounds were found to play a significant role in their DPPH and NO• radical scavenging properties.
A typical example of the EPR DPPH scavenging assay will be illustrated through a recent study conducted by our team, where the EPR DPPH scavenging assay was employed to measure the decay of the DPPH signal induced by six harmicenes (83). This was part of a broader investigation into their antiproliferative activity against a human malignant glioma cell line and the underlying mechanisms of action. The results of this experiment are presented in Fig. 10. which shows the reduction in the DPPH double integral value observed in the scavenging measurements of six distinct harmicenes. The area under the resonance absorption peaks (commonly referred to as the "double integral" in EPR spectroscopy) was used to quantify the remaining DPPH, as derived from the collected spectra.
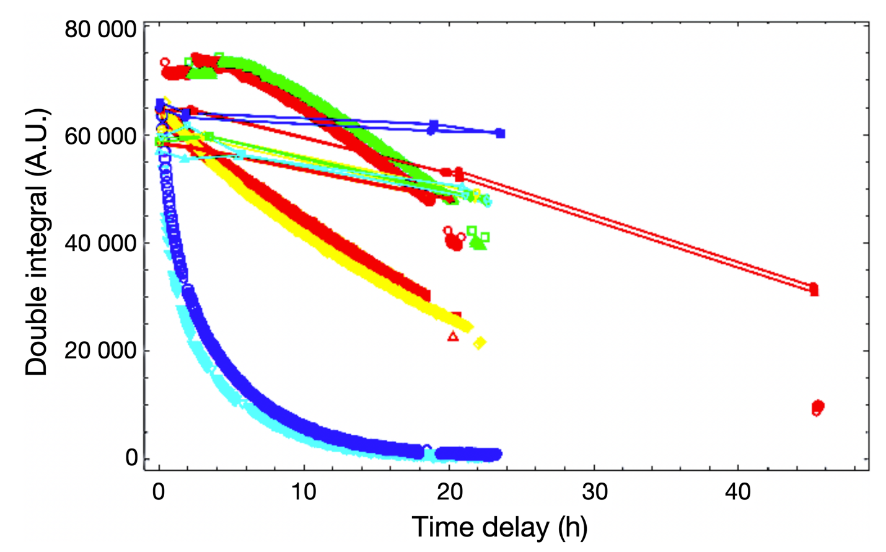
Fig. 10. The reduction in the DPPH double integral value observed in the measurements of a 1:1 mixture with six distinct harmicenes (each denoted by different colors). The solid lines connecting the small symbols represent the baseline measurements for each reaction mixture, corresponding to the colors used.
In conclusion, EPR spectroscopy provides exceptional sensitivity and specificity for analyzing the free radical scavenging properties of pharmaceutical compounds. Its application has been instrumental in characterizing the long-term stability and efficacy of antioxidants in pharmaceutical formulations. This versatility makes EPR spectroscopy a powerful tool for understanding the kinetics and efficiency of radical scavengers across a wide range of matrices.
APPLICATION OF EPR SPECTROSCOPY IN ELUCIDATING DRUG ACTIVITY, MECHANISMS OF ACTION, AND PHARMACOKINETICS
EPR spectroscopy is a highly effective analytical technique for pharmacokinetic studies involving paramagnetic compounds. Its primary advantage lies in its high specificity and the ability to detect paramagnetic analytes directly within biological samples, eliminating the need for sample preparation, purification, or separation. This capability significantly reduces analysis time and enhances reproducibility, which is crucial for analyzing highly reactive substances with very short half-lives that are sensitive to manipulation.
Beyond quantifying scavenging properties, EPR spectroscopy enables mechanistic studies that elucidate how pharmaceutical compounds neutralize free radicals. For instance, radical scavengers may act through electron transfer, hydrogen atom donation, or radical-radical recombination, each of which can manifest distinct EPR signatures (7, 8). By analyzing the spectra, the nature of the interactions between the scavenger and the radical can be inferred. Additionally, EPR spin labeling and spin trapping can be used to track the formation of intermediate species, providing further clarity on the stepwise process of radical neutralization (10). In more complex systems, such as drug-radical interactions in biological matrices, EPR spectroscopy is invaluable in isolating and characterizing multiple reaction pathways. For example, when pharmaceutical compounds target specific reactive oxygen species (ROS), EPR analysis can identify whether the compound preferentially scavenges superoxide over hydroxyl radicals, allowing for targeted drug design (84).
The integration of EPR spectroscopy with spin trapping techniques has been extensively applied to various classes of pharmaceutical compounds, including antihypertensives, antimalarials, antitrypanosomals, anticancer agents, vasodilators, and anti-ischemic drugs (85, 86). Furthermore, the DMPO spin trapping method has revealed that certain non-steroidal anti-inflammatory drugs, such as nimesulide and its principal metabolite 4-hydroxynimesulide, exhibit hydroxyl radical trapping and superoxide scavenging properties, which contribute to their anti-inflammatory and antirheumatic effects (87). Additionally, the calcium channel blocker nifedipine, known for its light sensitivity, undergoes photoreaction to form a nitroso compound, NTP, which displays spin trapping properties. The antioxidant efficacy of NTP was evaluated against lipid peroxidation in LDL and phosphatidylcholine liposomes and compared with other nitroso spin traps. NTP demonstrated superior antioxidant potency compared to nifedipine and other nitroso compounds (88).
It has been demonstrated that EPR studies have provided insights into the mechanism of a new anticancer agent iminoquinone, 5H-pyridophenoxazin-5-one, which acts through a free radical mechanism involving the formation of superoxide anions responsible for DNA cleavage (89). Fig. 11 shows the simulated EPR spectrum of the radical anion PPH•– formed by the reaction of the antitumor agent 5H-pyridophenoxazin-5-one with NADH in DMSO at room temperature. The cancer chemopreventive agent oltipraz has also been shown to generate superoxide anions in a concentration- and time-dependent manner, highlighting its role in chemoprevention (90). Moreover, EPR spin trapping has elucidated the mechanisms of peroxide antimalarials, such as artemisinin and dihydroartemisinin, revealing that these drugs produce C-radicals in the presence of haem iron, leading to the death of malaria parasites (91, 92). Fig. 12 presents the simulated EPR spectrum of the experimental one observed following the addition of human serum to an acetonitrile solution of artemisinin containing the spin trap DMPO. Similarly, DMPO spin trapping has been employed to investigate free radicals generated from nitrofuran derivatives of nifurtimox, correlating oxidative stress mechanisms with their anti-Chagas activity (93).
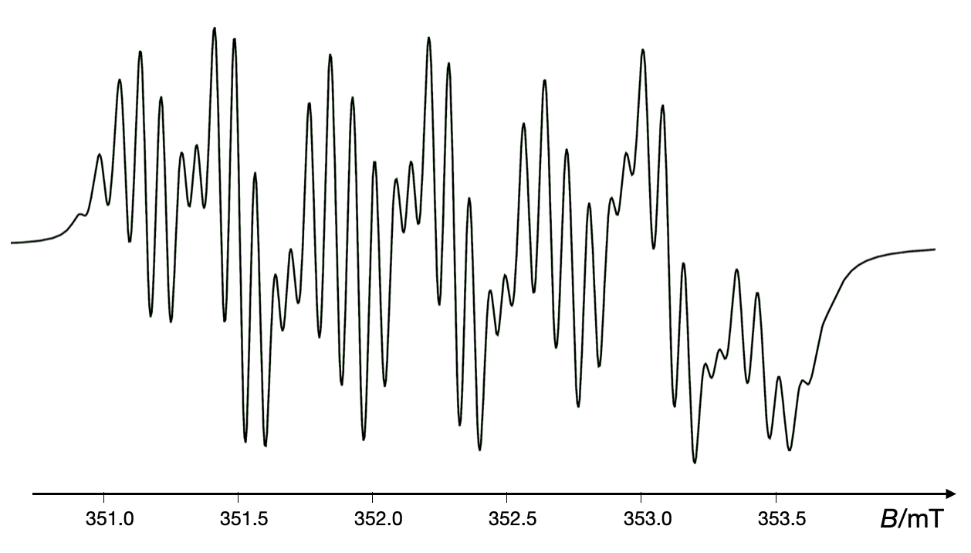
Fig. 11. Simulated EPR spectrum of the radical anion PPH•– formed by reaction of antitumor agent 5H-pyridophenoxazin-5-one with NADH in DMSO at room temperature. VisualEPR program was used to simulate the experimental data as reported in ref. 89.
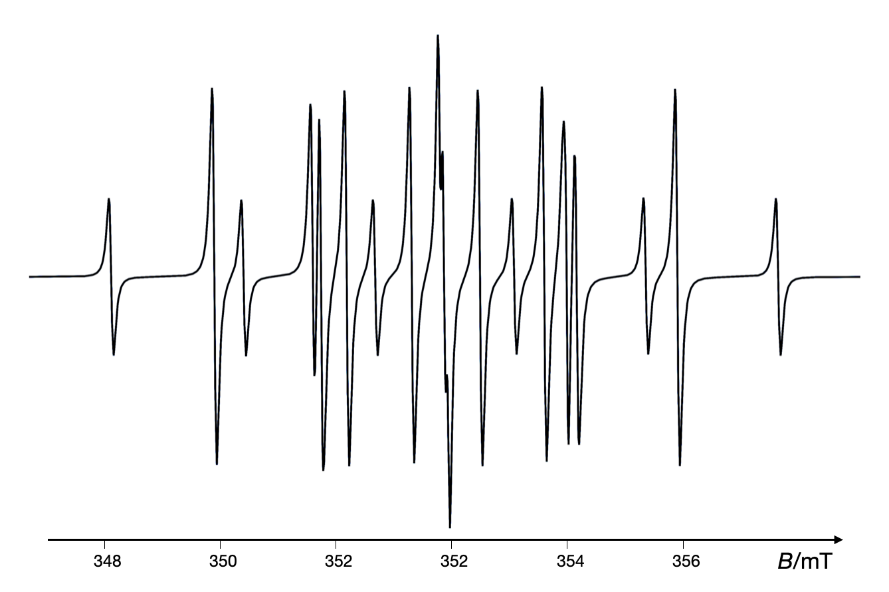
Fig. 12. Simulated EPR spectrum of the experimental one observed following the addition of human serum to an acetonitrile/H2O (1:1) solution of artemisinin containing spin trap DMPO at 37 °C, 50 minutes after serum addition. The spectrum was constructed by assuming the simultaneous presence of four types of nitroxides in the corresponding percentages. Simulated using the VisualEPR program based on data from ref. 92.
Although it exhibits lower sensitivity compared to advanced techniques such as liquid chromatography or gas chromatography coupled with mass spectrometry (LC-MS or GC-MS), EPR spectroscopy remains the method of choice for investigating paramagnetic species due to its unmatched ability to directly detect these analytes within complex biological matrices. This is exemplified by its application in researching insulinomimetic vanadyl VO2+ ions and their complexes, which are potential agents for diabetes prevention and treatment. Various oxovanadium(IV) complexes have been shown to normalize blood glucose levels in diabetic animal models (94).
Recent advancements include the development of an in vivo blood circulation monitoring method, BCM-EPR (95). A conventional X-band EPR spectrometer is utilized by this technique to detect stable free radicals in the bloodstream of anesthetized rats, allowing for real-time pharmacokinetic studies of stable spin probes. The method has been further employed to investigate the absorption, bioavailability, and blood disposition of various vanadyl complexes following intravenous or oral administration (94, 96). Furthermore, this approach has been used to assess the bioavailability of vanadyl compounds in enteric-coated capsules (97). Thus, EPR spectroscopy enables the detection of paramagnetic compounds in the blood and deep tissues, facilitating the characterization of drug absorption based on administration methods and the physical state of the delivery system (98). By analyzing changes in EPR signal intensity, researchers can gain insights into tissue perfusion and viability. Notably, nitroxides, which cannot cross cell membranes in normal tissues, exhibit significant reductions in EPR signal when injected, particularly in necrotic areas such as tumors. This property has led to the proposal of using nitroxides as selective necrosis MRI contrast agents to map necrotic regions within tumors in mouse models (99).
Further pharmacokinetic studies of nitroxides, such as carboxy-proxyl administered intravenously, involved inducing various pathological conditions in mice to evaluate the liver and kidney's roles in compound clearance. These conditions included restricted renal blood supply, acute hepatotoxin exposure, and lipopolysaccharide exposure. The pharmacokinetics was described using a two-compartment model, revealing that while the fast component was significantly altered in treated mice, the half-lives of the slow component remained largely unaffected (100, 101). EPR spectroscopy was also employed to investigate the impact of two aminoglycoside antibiotics, gentamycin and kanamycin, on the free radical characteristics of the biopolymer DOPA-melanin, which has a high content of o-semiquinone free radicals and has a strong binding capacity to different drugs as a protecting mechanism. The detection of a singular EPR signal in the analyzed samples suggests that the aminoglycoside antibiotics did not induce the formation of novel radical species within DOPA-melanin. The evaluation of free radical presence in both natural and synthetic melanins was similarly conducted through EPR analysis (102).
The application of EPR spectroscopy facilitated the examination of glucocorticosteroids, specifically betamethasone, clobetasol, and dexamethasone. The analysis focused on the extremely broadened line shape and parameters of the EPR spectra, recorded across a broad microwave power range. The results indicated that free radicals were generated in clobetasol and dexamethasone during the thermal sterilization process, whereas no free radicals were detected in thermally sterilized betamethasone (103). The optimal conditions for the sterilization of these drugs were determined based on the EPR findings. EPR spectroscopy has also been utilized to monitor manganese-based contrast agents' biodistribution, as demonstrated in studies evaluating Mn2+ encapsulated in liposomes and its clinical applications (104, 105). Additionally, in vivo EPR spectroscopy has proven to be effective in monitoring metal ion intoxication treatments. Another study has demonstrated the feasibility of detecting chromium(V) in vivo and monitoring treatment progress through the measurement of reductions in tissue chromium(V) levels (106).
In summary, EPR spectroscopy has proven to be a highly effective technique for pharmacokinetic studies of paramagnetic compounds, offering unmatched specificity and the ability to detect analytes directly in biological samples. EPR spectroscopy's ability to measure free radical scavenging, elucidate drug-radical interactions, and track intermediate species makes it invaluable for understanding drug mechanisms and their effects. Despite its lower sensitivity compared to other analytical methods, such as chromatography and mass spectrometry, EPR spectroscopy remains a preferred choice due to its direct and non-invasive, detection capabilities, which are crucial for real-time pharmacokinetic studies and exploring the biodistribution and metabolism of various compounds. Early applications of in vivo EPR spectroscopy focused on studying the distribution and metabolism of nitroxides, which were pivotal in advancing the technology and exploring its clinical utility, while modern applications use heavy metals and lanthanides for monitoring different conditions. The technique's recent advancements further enhance its potential for assessing drug pharmacokinetics and therapeutic efficacy. Additionally, in vivo EPR spectroscopy shows considerable promise for assessing the pharmacokinetics of paramagnetic drugs.
EMPLOYING EPR SPECTROSCOPY AS A METHODOLOGY FOR DEMONSTRATING THE ROLE OF FREE RADICALS IN DRUG-INDUCED TOXICITY AND DRUG-MEMBRANES INTERACTION
As mentioned before, free radicals are highly reactive molecules that can inflict oxidative damage on cellular macromolecules, including lipids, proteins, and DNA. In the context of drug-induced toxicity, these radicals often result from the metabolic activation of drugs into reactive intermediates (107). For example, acetaminophen metabolism leads to the formation of the highly reactive N-acetyl-p-benzoquinone imine (NAPQI), which can generate free radicals and cause liver damage (108). The sensitivity of EPR spectroscopy allows for the precise measurement of these radical species, thereby facilitating an in-depth investigation into their roles in drug-induced oxidative stress. By capturing the presence of free radicals that other methods might miss, EPR spectroscopy provides crucial data on the extent of oxidative damage and its correlation with drug toxicity.
Free radical intermediates, in addition to contributing to the therapeutic mechanisms of various drugs, are also implicated in the manifestation of adverse drug reactions. For instance, several drugs, such as anthracycline quinones (e.g., adriamycin, ADM), anthralin derivatives, and enzymes like nicotinamide adenine dinucleotide phosphate (NADPH) P450 reductase and mitochondrial NADH dehydrogenase, exhibit free radical activity (109). Upon interaction with oxygen, reductive semiquinone radicals generate superoxide and hydroxyl radicals, initiating lipid peroxidation in unsaturated membrane lipids, which consequently leads to membrane destabilization and mitochondrial dysfunction, particularly in cardiac cells (110). Notably, ADM-induced cardiotoxicity was among the first examples where EPR techniques were employed to elucidate free radical-mediated drug toxicity mechanisms (111).
Antimalarial and non-steroidal anti-inflammatory drugs (NSAIDs) also demonstrate significant free radical involvement in their side effects. EPR studies have provided insights into primaquine-induced hemolytic anemia, revealing that the N-hydroxy metabolite of primaquine, 6-methoxy-8-hydroxylaminoquinoline (MAQ-NOH), acts as a direct hemolytic agent. Spin trapping techniques have confirmed that oxygen-derived radicals play a central role in the drug's hemolytic damage (112).
Phototoxicity is another free radical-associated side effect of several antimalarial drugs, including primaquine, chloroquine, and quinacrine. Upon UV exposure, these drugs generate reactive intermediates, including superoxide and hydroperoxyl radicals, as demonstrated by the DMPO spin trapping technique (113). The prevalence of malaria in high-intensity light regions suggests that protective measures should be considered during treatment with these drugs to mitigate phototoxic reactions.
In the context of NSAID-induced gastric mucosal damage, EPR studies have demonstrated that indomethacin (IM) radicals generated through interaction with horseradish peroxidase and hydrogen peroxide are involved in lipid peroxidation and sulfhydryl enzyme inactivation (114). These radicals contribute to cellular damage via mechanisms involving the reduction of iron to ferryl species, followed by superoxide anion formation. Similarly, piroxicam, another NSAID, has been shown to inactivate alcohol dehydrogenase through free radical interactions, with glutathionyl radicals playing a crucial role in this process (115, 116). These findings suggest that lipid peroxidation and enzyme inactivation are key factors underlying the tissue damage associated with NSAID usage. Fig. 13 illustrates simulated EPR spectra of glutathionyl radical formation by piroxicam in the presence of horseradish peroxidase (HRP), hydrogen peroxide (H₂O₂), and reduced glutathione (GSH), investigated under eight different conditions based on variations of the reaction mixture.
Antiandrogen nilutamide, utilized in the treatment of metastatic prostate carcinoma, is associated with adverse reactions such as acute hepatitis and pulmonary fibrosis (117). EPR studies have shown that nilutamide undergoes one-electron reduction via NADPH-cytochrome P450 reductase, generating a nitroanion free radical, which under anaerobic conditions forms reactive species that covalently bind to cellular macromolecules. Under aerobic conditions, the nitroanion free radical engages in a redox cycle, producing an excess of reactive oxygen species (ROS), including superoxide anion and hydrogen peroxide, which contribute to the drug's toxicity in hepatocytes (118).
EPR spectroscopy has also been instrumental in the non-invasive monitoring of free radical generation in vivo, as exemplified by the detection of radical formation in the skin following the topical application of anthralin. This was observed under therapeutic conditions, where the radicals were linked to the metabolic byproducts of anthralin (119).
In addition, EPR spin trapping has recently been applied to improve the photostability of sunscreen agents such as phenylbenzimidazole sulphonic acid (120). The inclusion of this UV-B filter into hydroxypropyl-β-cyclodextrin was found to significantly suppress the generation of light-induced free radicals, thereby reducing potential DNA damage caused by solar-simulated radiation (121). These results underscore the utility of EPR techniques in both understanding drug-induced toxicity and enhancing drug and chemical stability through free radical modulation.
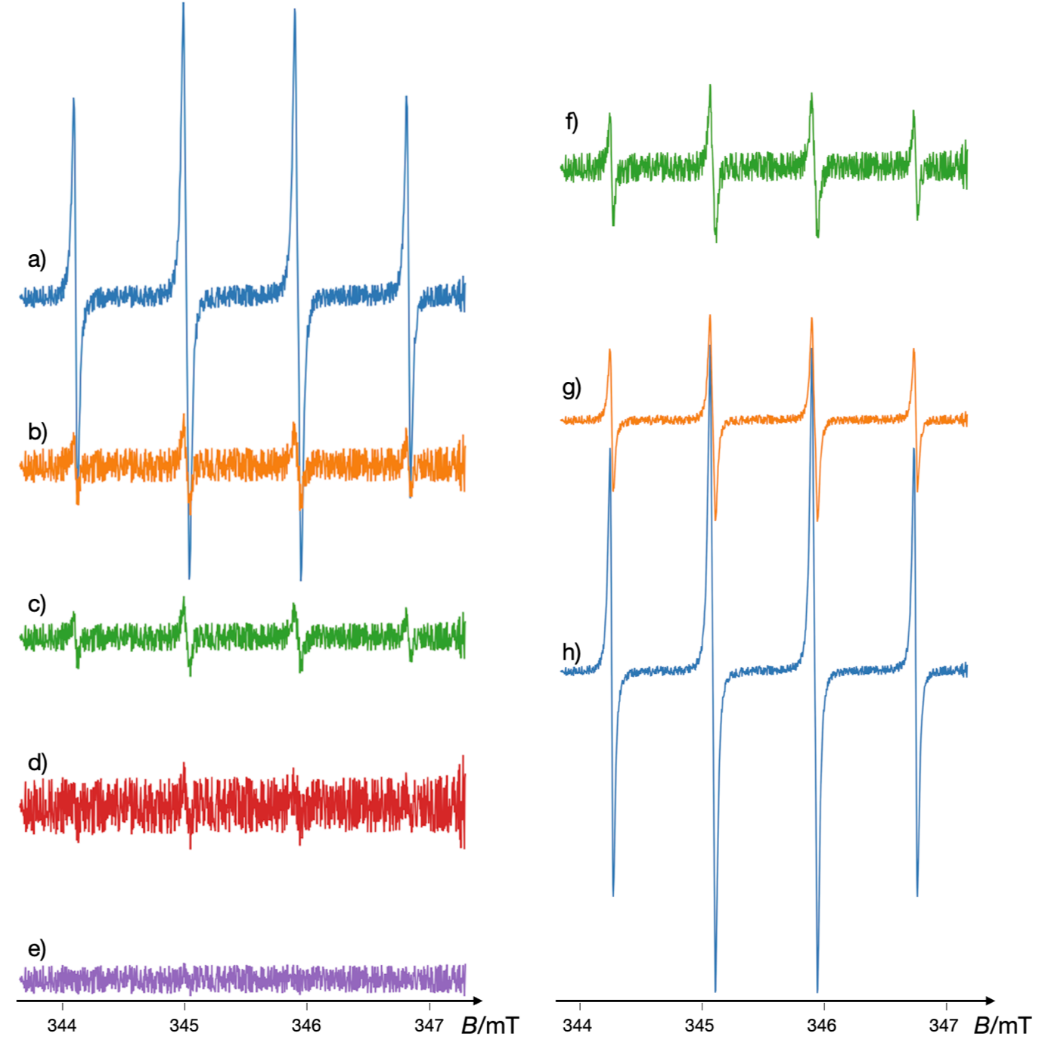
Fig. 13. Simulated EPR spectra (with added noise) of the formation of glutathionyl radicals by piroxicam in the presence of horseradish peroxidase (HRP), hydrogen peroxide (H2O2), and reduced glutathione (GSH) investigated under the following conditions: a) complete reaction mixture; b) without piroxicam; c) without H2O2; d) without GSH; e) without HRP; f) reaction mixture with heat-denatured HRP; g) complete reaction mixture with superoxide dismutase (SOD); h) complete reaction mixture under anaerobic conditions. Simulated using VisualEPR program with data from ref. 119.
The detection and quantification of drug effects on cellular membranes are crucial for understanding drug action during drug development. A variety of methodologies are employed to investigate the interactions between drugs and membrane properties, including high-performance liquid chromatography (HPLC), differential scanning calorimetry (DSC), fluorescence spectroscopy, Fourier transform infrared spectroscopy (FT-IR), X-ray crystallography, nuclear magnetic resonance (NMR), circular dichroism (CD), and their combinations. Since the 1960s, EPR spectroscopy has been utilized to explore the interactions of psychotropic drugs, such as chlorpromazine, promazine, and perphenazine, with cell membranes using spin-labeled erythrocyte ghosts (122). A notable example is the investigation of the NSAID sulindac and its metabolites (sulfide and sulfone) with model membranes having different transition temperatures, such as dispersed vesicles and multilamellar liposomes of dioleoyl-L-α-phosphatidylcholine and dipalmitoyl-L-α-phosphatidylcholine (123, 124). In these studies, the perturbation of lipid bilayer dynamics induced by drug interactions was assessed using spin probes like 2,2,6,6-tetramethylpiperidinyl-1-oxy (TEMPO) and C5- and C12-doxylstearic acid.
Studies since the 1980s have demonstrated the utility of spin label EPR spectroscopy for analyzing various membrane properties, including lipid phase transitions, membrane permeability, and lipid-protein interactions (125, 126). For example, spin-labeled stearic acid was used to evaluate the effects of local anesthetics, such as tetracaine and dibucaine, on synaptosomes, and to assess the influence of β-adrenoceptor blockers on synaptosomal membranes (127, 128). Fig. 14 shows the simulated EPR spectra of the stearic acid spin label (16-SASL) in synaptosomes treated with three different beta-blockers (atenolol, doberal, and propranolol), compared with the control.
Additionally, spin probes like 16-doxyl stearic acid and 14-doxyl phosphatidylcholine were employed to compare the perturbation effects of propranolol, verapamil, chlorpromazine, and carbisocaine on lecithin and brain lipid liposomes (129). Findings from these studies indicate that β-adrenoceptor blockers increase liposome disorder in proportion to their antiaggregatory effects, while no similar relationship was observed for calcium channel blockers. Other studies have explored the interactions of modafinil and amphetamine with membrane models, including small and large unilamellar vesicles, using stearic acid spin label probes (130). The results revealed distinct fluidity gradients induced by these drugs, which could explain the differing pharmacological potencies of amphetamine and modafinil.
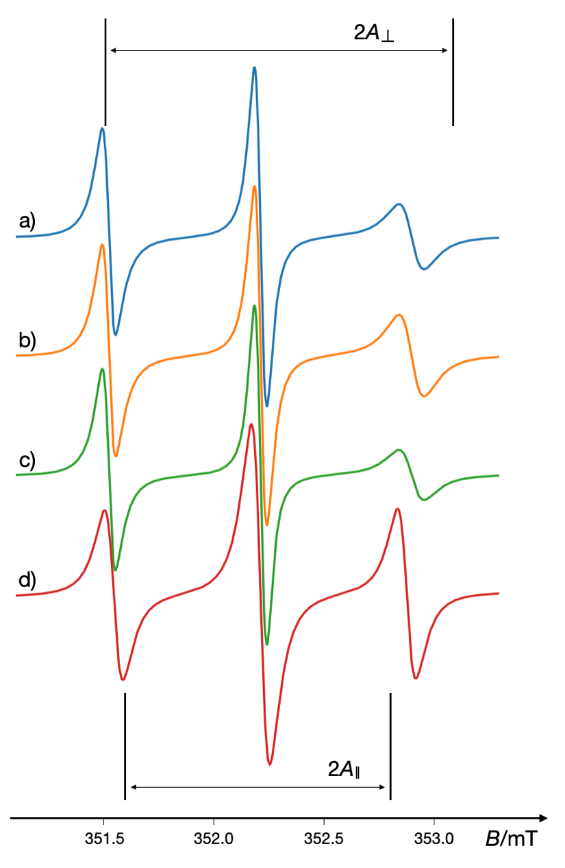
Fig. 14. Simulated EPR spectra of the stearic acid spin label (C12-doxylstearic acid, 16-SASL) in the synaptosome treated with: a) atenolol; b) doberal; c) propranolol and d) control sample measured at 37 °C. An increase of parameter A⊥, and a decrease of parameter A∥ indicates higher disorder or dynamics of the hydrophobic part of lipid membranes. The results indicate that drugs had different effects on the dynamics and/or order of the hydrophobic part of the bulk lipid in the synaptosomal membranes. Simulated using VisualEPR program with data from ref. 128.
Alkylphospholipids, such as octadecyl-(1,1-dimethyl-4-piperidino-4-yl)-phosphate (OPP), have shown significant antitumor activity in liposome suspensions with low cholesterol content. EPR studies using 5-doxylpalmitoyl methyl ester as a spin probe demonstrated that the therapeutic efficacy of alkylphospholipid formulations is influenced by the cholesterol-to-OPP ratio, indicating the importance of membrane fluidity in their therapeutic activity (131). EPR studies with spin-labeled fatty acids and phosphatidylcholine have provided valuable information on the distribution of these probes in solid lipid nanoparticles (SLNs) and their interactions with biological membranes (132). EPR analyses revealed that smaller spin probes exhibited faster passive transport across model membranes, though similar transfer rates were observed for SLNs and leukocytes, suggesting potential endocytic uptake of SLNs. Recent investigations using EPR spectroscopy have highlighted the potential of magnetically aligned phospholipid bilayers, or bicelles, as model membrane systems. The dynamic properties of 1,2-dimyristoyl-sn-glycero-3-phosphocholine/1,2-dihexanoyl-sn-glycero-3-phosphocholine (DMPC/DHPC) bicelles were found to align with other biological and model membranes, confirming their suitability for various biophysical applications (133).
In summary, free radicals play a dual role in drug action and toxicity, with their generation often resulting from the metabolic activation of drugs into reactive intermediates. Techniques such as EPR spectroscopy are crucial for detecting and quantifying these radicals, providing insights into oxidative damage and drug-induced toxicities. EPR spectroscopy has revealed the mechanisms behind drug-related adverse effects, including oxidative stress and phototoxicity, and has been instrumental in developing strategies to mitigate these effects. Furthermore, EPR's applications extend to understanding drug-membrane interactions and optimizing drug formulations, showcasing its versatility in drug development and safety evaluation.
APPLICATION OF EPR SPECTROSCOPY IN DRUG DELIVERY RESEARCH
EPR spectroscopy offers a powerful framework for advancing drug delivery research, particularly in monitoring delivery mechanisms both in vivo and in vitro. This method has proven to be highly effective in the characterization and monitoring of various types of drug carriers, including colloidal, non-transparent, and solid systems, providing detailed insights into their physicochemical properties such as stability, charge, size, and mobility (134). These characteristics are critical in drug delivery carriers like liposomes, micelles, nanoparticles, dendrimers, cyclodextrins, and cucurbiturils, which have been widely used to transport therapeutic agents to specific tissues and organs (135). EPR spectroscopy, therefore, plays a significant role in assessing the structural dynamics and functional performance of these drug delivery vehicles.
A primary strength of EPR spectroscopy lies in its capacity to provide real-time insights into the dynamics of drug release mechanisms. The technique allows for the measurement of key parameters, including nitroxide concentration, micro-polarity, micro-viscosity, and micro-acidity, through distinct spectroscopic signatures (136). For example, the nitroxide concentration can be quantified by double integration of the EPR spectra, while micro-polarity and micro-viscosity are determined through the hyperfine coupling constants and the shape of the spectra, respectively (136). These measurements are crucial in ensuring that the release mechanisms observed in vitro are reflective of the in vivo environment. EPR spectroscopy has been particularly effective in identifying discrepancies in drug release between these two contexts due to variations in water accessibility, pH, and enzymatic activity, which can significantly influence drug stability and efficacy (137). Such discrepancies underscore the importance of employing EPR spectroscopy to thoroughly characterize drug delivery systems across both experimental conditions.
Furthermore, EPR spectroscopy is invaluable in studying the intricate details of drug release from complex carriers such as biodegradable polyesters and polyanhydrides. These systems exhibit time-dependent release profiles, often influenced by factors such as microviscosity, microacidity, and the local environment within the carrier (138–140). For instance, the ability to measure microviscosity and other critical parameters inside the carrier through EPR spectroscopy ensures a more comprehensive understanding of drug release kinetics and mechanisms, especially for drugs of different molecular weights, such as peptides and proteins, where spin labeling may be required (141).
Employing EPR spectroscopy enables the simultaneous measurement of microviscosity and micropolarity in the oil and aqueous phases of oil-in-water or water-in-oil dispersions can be achieved with great rapidity by employing a nitroxide probe that is capable of partitioning across both phases. This technique proves invaluable for the analysis of samples that are typically challenging to assess, such as nontransparent, solid, multiphase, or nanometer-scale heterogeneous samples. These methodologies hold particular significance in the study of colloidal drug delivery systems, which, owing to their submicron size, are often difficult to evaluate, with many being sensitive to dilution. Detailed investigations have elucidated the effects of lipid composition and polyethylene glycol (PEG) chain length on the properties of PEGylated liposomes (142). Additionally, a study investigating the interactions between colloidal lipid dispersions and cells has demonstrated that the transfer is significantly influenced by the spin probe, colloid matrix, and surfactant employed (132).
An illustrative example of EPR application in colloidal systems is depicted in Fig. 15, demonstrating that a low-concentration vesicular lipid dispersion (2 %) can encapsulate up to 94 % of the lipophilic model drug, tempolbenzoate (TB) (137). However, it has recently been discovered that colloidal solid lipid dispersions possess limited loading capacity, primarily due to the crystalline nature of the lipid components. Comparative analyses of solid lipid nanoparticles (SLN) and nanostructured lipid carriers (NLC), the latter comprising both solid and liquid lipids, have shown that these systems do not exhibit significant advantages over submicron emulsions. This limitation arises from lipid crystallization, which causes the expulsion of the lipophilic nitroxide into the aqueous phase, as evidenced by approximately two-thirds of the TB being exposed to a polar environment in a 10 % colloidal dispersion of solid lipids (36). It is widely acknowledged that the surface properties of nanoparticles are a critical determinant of their biological fate. For example, a study employing EPR spectroscopy has elucidated that the surface properties of poly(isobutylcyanoacrylate) nanoparticles are contingent upon the specific synthetic method used (143).
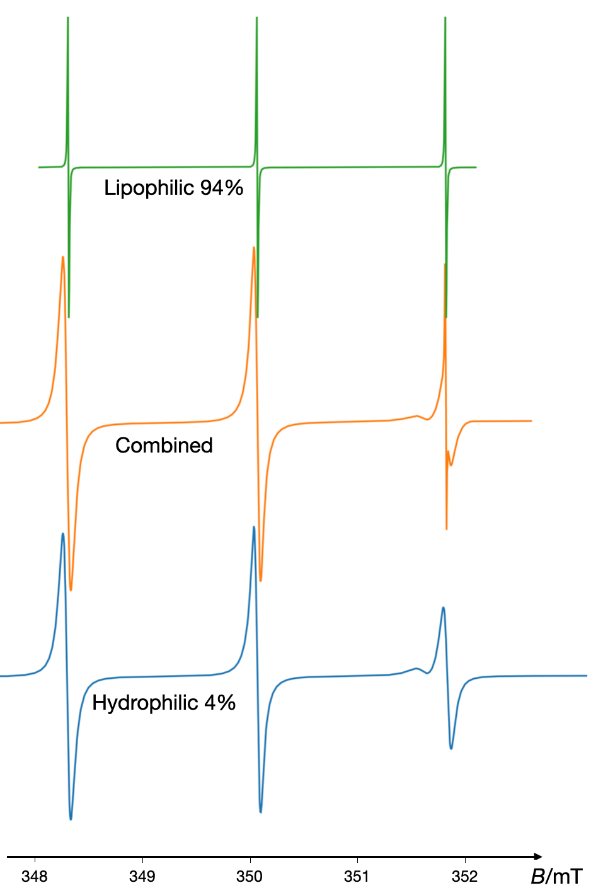
Fig. 15. The EPR spectrum of lipophilic spin probe Tempolbenzoate (TB) in low concentrated colloidal dispersion of amphiphilic sucrose esters. Quantitative analysis using a combination of two simulations indicates that 94 % of TB is localized in a lipophilic environment with increased viscosity and 4 % is localized in a low viscous, polar environment. The simulation was performed using the VisualEPR program, drawing on data from ref. 137.
The protective capabilities and distribution dynamics of colloidal drug carriers are crucial for their overall performance, yet these parameters are often difficult to quantify. They can, however, be evaluated through assays involving ascorbic acid reduction or dilution (144, 145). Hydrophilic ascorbic acid rapidly reduces piperidine nitroxides to their EPR-silent hydroxylamine forms, with the subsequent loss of EPR signal intensity indicating the degree of nitroxide accessibility to the outer aqueous phase. In the dilution assay, the spectral shape changes in response to the incremental addition of the outer phase, providing insights into distribution processes. Current research involving various colloidal carriers, such as solid lipid nanoparticles, nanoemulsions, nanocapsules, and polymeric nanoparticles, suggests that distribution processes occur within a timespan of seconds to minutes. This is expected given the short diffusion distances within the particles and the minimal thickness (e.g., 10 nm) of nanocapsule shells (144, 145). It is worth noting that solid colloidal drug carriers are exceedingly difficult to study using conventional NMR, and even solid-state NMR encounters challenges due to their short relaxation times and artifacts resulting from the high shear stress induced by rapid sample spinning.
Beyond characterizing drug delivery systems before their application, EPR spectroscopy also provides critical insights into the release mechanisms of drugs. The technique offers unique and essential information, as distinct release mechanisms induce specific alterations in spectral intensity and shape, as illustrated in Fig. 16. In a purely erosion-controlled release mechanism, the EPR signal intensity decreases over time, but the spectral shape remains consistent throughout the release period. Conversely, diffusion and swelling-controlled release mechanisms cause significant spectral shape changes, owing to the contribution of solubilized, rapidly tumbling nitroxide. Likewise, EPR spectroscopy has been extensively employed to monitor release processes from matrices such as hydroxypropyl methylcellulose (HPMC) and albumin, providing detailed insights into the impact of incorporated drugs on these release dynamics (146, 147).
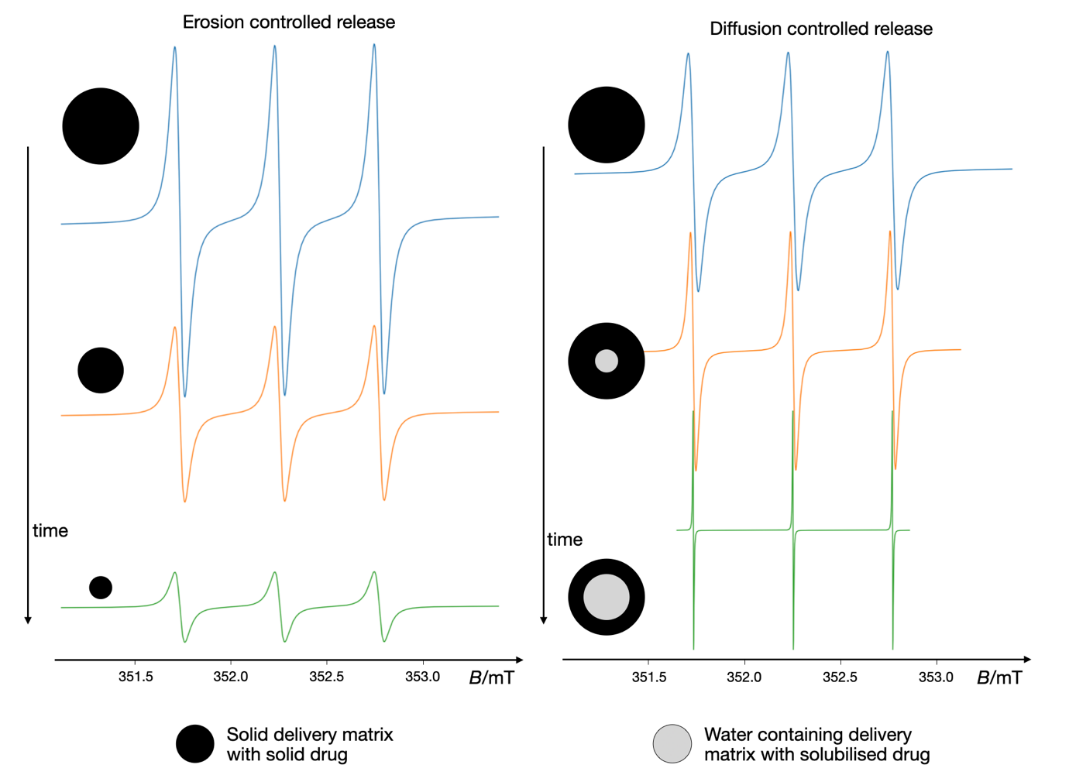
Fig. 16. Monitoring drug release mechanisms by EPR spectroscopy – the release of incorporated nitroxide in pure erosion-controlled release (characterized by a decrease in signal intensity without spectral shape changes) vs. diffusion and swelling-controlled release (where water penetration leads to solubilization and narrowing of the spectral lines). Quantification of solubilized nitroxides (three-line spectrum with narrow lines) and immobilized nitroxides can be achieved through spectral simulation and double-integral calculations. The simulation was executed with the VisualEPR program, incorporating data from ref. 137.
A particularly important application of EPR spectroscopy in drug delivery is the study of microacidity within drug carriers, a factor that significantly impacts drug solubility, polymer degradation, and monomer clearance. EPR spectroscopy is highly sensitive to local pH changes, allowing for the detection of pH gradients within drug carriers and providing crucial insights into how these gradients affect the release and stability of the drug (148). Studies have demonstrated that spatial-EPR-imaging can detect pH gradients on the order of 2 pH units within a few micrometers inside drug carriers, although achieving this resolution in vivo remains a challenge (148, 149). Such precise pH monitoring is vital for the optimization of pH-sensitive drug delivery systems, as the microenvironment within the carrier can directly influence the drug’s pharmacokinetics and therapeutic efficacy.
The application of EPR spectroscopy also extends to the imaging and spatial mapping of drug carriers. EPR imaging has made it possible to visualize the distribution of drugs within biological tissues, enabling the concurrent monitoring of both the carrier and the drug, particularly when using different nitroxide isotopes for labeling (16, 150). This is exemplified in studies where 2D and 1D EPR imaging techniques were used to map the distribution of liposome-encapsulated drugs in living systems. For example, nitroxide-labeled liposomal formulations were injected into the femoral muscle of a living mouse, and the EPR signal from both encapsulated and free drug molecules was separately reconstructed, allowing for precise mapping of drug localization (151). Such imaging capabilities are indispensable for studying the release dynamics and biodistribution of drugs in vivo, especially for carriers like liposomes and nanoparticles, which are often used in advanced drug delivery systems.
Despite its numerous advantages, EPR spectroscopy does face certain limitations, particularly in the context of in vivo studies. Penetration depth and sensitivity remain major challenges, especially for long-term drug release systems, where signal attenuation can occur over extended periods (16, 152). Additionally, most drug delivery systems are EPR-silent due to the absence of paramagnetic species. While the addition of paramagnetic materials, such as stable organic radicals (e.g., nitroxides), allows for enhanced detection and analysis, it introduces complexities that must be carefully managed. These paramagnetic probes, while invaluable in studying the microenvironment of drug carriers, can sometimes alter the native behavior of the drug delivery system. Nonetheless, the inclusion of nitroxides as model drugs or spin probes remains a common and effective strategy in EPR studies of drug delivery (153).
Recent advances in the development of novel drug delivery systems have benefited greatly from EPR analysis. For instance, chimeric advanced drug delivery nanosystems and PEGylated liposomal formulations have been extensively studied using EPR spectroscopy. In a notable study, liposomes comprising a blend of phosphatidylcholines and hyper-branched polymers were analyzed, and EPR spectroscopy demonstrated that these formulations exhibited enhanced stability and improved drug release profiles due to the presence of the hyper-branched polymers (154). Moreover, PEGylation was shown to further stabilize the drug within the liposome, offering a promising approach for increasing the therapeutic efficacy of drugs such as shikonin (154). This type of analysis underscores the critical role of EPR spectroscopy in refining drug delivery formulations, ensuring that they maintain stability and controlled release under physiological conditions.
To conclude, EPR spectroscopy remains a cornerstone technique in the field of drug delivery research, providing unparalleled insights into the structural and dynamic properties of drug carriers. Its ability to monitor key parameters such as microviscosity, micropolarity, and microacidity, along with its imaging capabilities, makes it indispensable for the in-depth analysis of drug release mechanisms. While challenges such as penetration depth and sensitivity persist, ongoing advancements in EPR technology and methodology continue to enhance its utility in both in vitro and in vivo studies. By facilitating a more comprehensive understanding of drug delivery systems, EPR spectroscopy not only aids in the optimization of existing formulations but also paves the way for the development of innovative therapeutic platforms capable of addressing the complex demands of modern medicine.
ADVANCED APPLICATIONS OF EPR SPECTROSCOPY IN THE STUDY OF METALLODRUGS
EPR spectroscopy has emerged as a powerful analytical technique for investigating the structural, dynamic, and mechanistic properties of metallodrugs (26), a class of therapeutic agents that contain metal ions as integral components. Metallodrugs are increasingly recognized for their diverse biomedical applications, including their use as anticancer, antimicrobial, and antiviral agents, due to the unique properties imparted by metal centers (155). The paramagnetic properties inherent to numerous transition metals and their complexes make EPR spectroscopy an exceptionally effective tool for investigating metallodrugs. This technique provides valuable insights into their coordination environments, redox states, conformational and structural alterations, ligand-exchange dynamics, pH-dependent behavior, redox mechanisms, metal coordination with organic ligands, and interactions with biological targets (26, 28).
One of the primary advantages of EPR spectroscopy in metallodrug research is its ability to provide detailed information about the local environment of unpaired electrons associated with metal centers, such as in complexes of copper(II), iron(III), and platinum(II) (24, 26, 156). EPR spectroscopy can distinguish between different oxidation states of metal ions, elucidate ligand coordination geometries, and monitor changes in the electronic structure of metallodrugs during biological interactions. For example, studies on copper-based metallodrugs have leveraged EPR spectroscopy to investigate the binding of copper(II) ions with ligands and their redox behavior in cellular environments (157). Such insights are invaluable for understanding the mechanisms by which metallodrugs exert their therapeutic effects, particularly in redox-active systems.
The structural characterization of anticancer metal complexes, especially when comparing their solid-state forms to their behavior in solution, is essential in understanding their potential therapeutic effects. For instance, X-ray crystallography often depicts bonding configurations that may differ from the structures found in aqueous solutions, where the complexes are typically administered. EPR spectroscopy has been pivotal in resolving such discrepancies, particularly in distinguishing between monomeric and dimeric species based on spin coupling effects. A notable example is the study of cytotoxic Cu(II)-proline-thiosemicarbazone complexes. EPR spectra at low pH show a monomeric Cu(II) species, while at higher pH, the spectrum reveals a second species with distinctive g-values and a half-field peak diagnostic of a spin-coupled system, indicating the presence of a dimeric Cu(II) species (158). Similar EPR investigations have demonstrated that some dimeric complexes observed in the solid state dissociate into monomeric species in solution, as seen in cytotoxic Cu(II)-quinolone complexes (159) and pyrazole-based ligands (160, 161). Additionally, EPR spectroscopy has probed isomerization in solution, as shown by a study on an antitumor oxovanadium complex, where two isomers were identified based on differences in their hyperfine splitting patterns, relevant to the complex's cytotoxicity through DNA intercalation (162).
Moreover, EPR spectroscopy has been instrumental in characterizing the interactions between metallodrugs and biomolecules, including proteins, nucleic acids, and lipids. The application of spin labeling techniques in EPR spectroscopy allows for the identification of metallodrug binding sites and provides information on the drug's mode of action at the molecular level (163). For instance, EPR studies have revealed that platinum-based drugs, such as cisplatin, form covalent adducts with DNA, leading to cross-linking that disrupts cellular replication and promotes apoptosis (164). EPR spectroscopy has been especially useful in studying metal coordination by organic therapeutic agents, such as the anticancer drug bleomycin (165). The activity of bleomycin is linked to its coordination with redox-active metals, particularly Fe(III), which can be sourced from the biological environment. EPR and electron-nuclear double resonance (ENDOR) studies have characterized the Fe(III)-bleomycin complex, particularly its interactions with oxygen, identifying the Fe(III)-bleomycin hydroperoxide species as the active form responsible for DNA strand cleavage (166, 167). The coordination of oxygen was confirmed through EPR studies using 17O2, and the mechanism of DNA cleavage was further validated by changes in the EPR spectrum in the presence of DNA (168). Bleomycin also forms active complexes with Cu(II) and Co(II), which have been similarly characterized by EPR (169).
Halouracils and their derivatives, such as 5-fluorouracil, have demonstrated notable antitumor and antiviral properties, with 5-fluorouracil being widely used for the treatment of various cancers (170, 171). Other halouracils, like 5-chlorouracil and 5-bromouracil, have shown potential in treating inflammatory conditions (172). Complexes of 5-iodouracil with transition metal ions, particularly redox-active metals like copper, cobalt, and zinc, exhibit strong DNA binding and induce oxidative DNA damage, making them promising candidates for anticancer therapies (173, 174). Substituted 6-xuracils (X F, Cl, Br, and I) have also been investigated as potential inhibitors of nucleic acid metabolism and as clinical radiosensitizers for DNA in tumor cells, aiming to mitigate damage to healthy tissues during radiation therapy (175). In our recent study, we conducted an extensive single-crystal EPR analysis to investigate the paramagnetic complex formed between Cu(II) ions and 6-chlorouracil molecules within a single crystal of 6-chlorouracil containing copper impurities (176). Fig. 17 presents a highly intricate EPR spectrum of this paramagnetic complex, comprising four groups of resonance lines that exhibit an additional quintet super-hyperfine structure arising from two copper isotopes. The precise spatial arrangement of the copper ions within the host lattice was elucidated, alongside a detailed exploration of the mechanism underlying the formation of the Cu(II)-6-chlorouracil paramagnetic complex induced by gamma-irradiation.
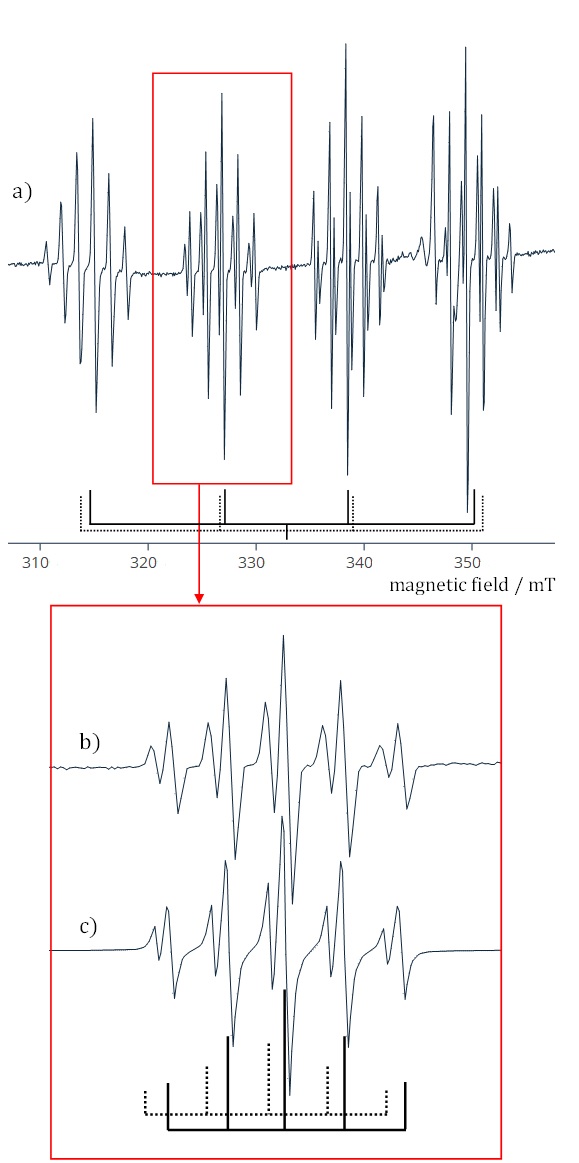
Fig. 17. a) EPR spectrum of paramagnetic complex Cu(II)-6-chlorouracil. The bars under the recording indicate the positions of four groups of lines for two distinct spectra, associated with 63Cu (solid lines) and 65Cu (dashed lines) isotopes; b) the super-hyperfine structure of the second spectral group of lines from Fig. 2a, as a result of hyperfine interaction with two magnetically equivalent nitrogen nuclei); c) the second group of lines from Fig. 2a simulated using the VisualEPR program (177).
In addition to characterizing interactions between metallodrugs and biomolecules, EPR spectroscopy has proven highly effective in monitoring ligand-exchange processes in metallodrugs, which often involve exchangeable ligands being replaced under physiological conditions. This is particularly important for prodrugs, which become active following ligand exchange in vivo (178). For paramagnetic metallodrugs, EPR spectroscopy directly reflects changes in the coordination environment of the metal center, enabling precise monitoring of these processes. Prime examples are the Ru(III) anticancer complexes KP1019, NKP-1339, and NAMI-A. These compounds undergo slow ligand-exchange processes in aqueous environments, which have been extensively studied through EPR spectroscopy (179, 180). EPR spectra from frozen solutions of these complexes show distinct signals from Ru(III) centers, which can be deconvoluted using spectral simulation to track the formation of multiple species during ligand exchange (181, 182). In some cases, ligand exchange in aqueous solutions leads to the formation of EPR-silent species due to the generation of multinuclear species linked through oxo or hydroxo bridges, such as in Ru(III) complexes, where antiferromagnetic coupling results in species (183). EPR spectroscopy has also suggested that the substitution of donor ligands can inhibit the formation of these multinuclear species, preserving the paramagnetic properties of the complexes (184).
EPR spectroscopy can sensitively report on pH-dependent speciation of metallodrugs, providing insights into their behavior in different biological environments. For example, many tumors exhibit a lower pH (pH5.8) compared to the bloodstream (pH = 7.4), and this variation can significantly influence the speciation of metal-based drugs (185). EPR spectroscopy has been used extensively to study pH-effects on Cu(II) and V(IV) thiosemicarbazones (158, 186, 187), revealing up to eight different species in solution. The simulations of EPR spectra enabled the identification of various species that predominated at different pH levels, with the major species at neutral pH being mono-ligand copper(II) complexes, and protonation or deprotonation leading to the formation of other species. Stability constants for the precipitated bis-ligand complexes [CuL2] could be only analyzed with EPR spectroscopy (186). This precise characterization of species across a pH range highlights the utility of EPR in constructing concentration distribution curves that are vital for understanding the biological activity of these complexes.
In conclusion, EPR spectroscopy has proven to be an indispensable tool in the investigation of metallodrugs, offering comprehensive insights into their structural dynamics, ligand-exchange processes, redox behavior, and metal coordination. This technique has notably enhanced our understanding of the mechanisms underlying metallodrug activity, particularly in oncology and antitumor therapy, while also providing valuable pathways for optimizing their therapeutic potential. The ability of EPR to examine both molecular-level drug interactions and broader pharmacokinetic properties underscores its critical role in advancing medicinal chemistry and facilitating the development of more effective therapeutic agents.
MEASURING OXYGEN CONCENTRATION IN BIOLOGICAL AND CHEMICAL SYSTEMS VIA EPR OXIMETRY
EPR oximetry is a sophisticated technique that measures oxygen concentrations in biological and chemical systems using EPR spectroscopy. This method leverages the use of paramagnetic materials that respond to variations in oxygen levels by altering their EPR spectral characteristics. When these materials are exposed to a magnetic field, they absorb microwave radiation at specific frequencies. The presence of oxygen influences the energy levels of the unpaired electrons, thereby modifying the EPR spectrum. By analyzing these spectral changes, researchers can accurately quantify the concentration of oxygen in the sample.
In practice, EPR oximetry employs a range of oxygen-sensitive materials. These include soluble radicals, such as nitroxides and their derivatives, which may be dissolved in solutions or encapsulated in carriers like liposomes or nanoparticles. The spin-label probe 3-carbamoyl-2,2,5,5-tetramethyl-3-pyrroline-1-yloxyl (CTPO) has demonstrated considerable efficacy in determining the concentration of dissolved oxygen in a variety of organo-chemical and biological samples, particularly in aqueous environments (188). In oxygen-free conditions, the rapid tumbling of this small probe averages out anisotropic magnetic interactions, resulting in a well-resolved EPR spectrum characterized by a distinct superhyperfine structure, which typically consists of twelve resonance lines. This resolution deteriorates as oxygen is introduced into the sample, due to molecular collisions between oxygen and the spin label, which leads to broadening of the resonance lines. Fig. 18 displays the twelve-line motif observed in the central resonance line of the experimentally obtained (a) and simulated (b) EPR spectrum of the spin probe CTPO in fully deoxygenated toluene (189).
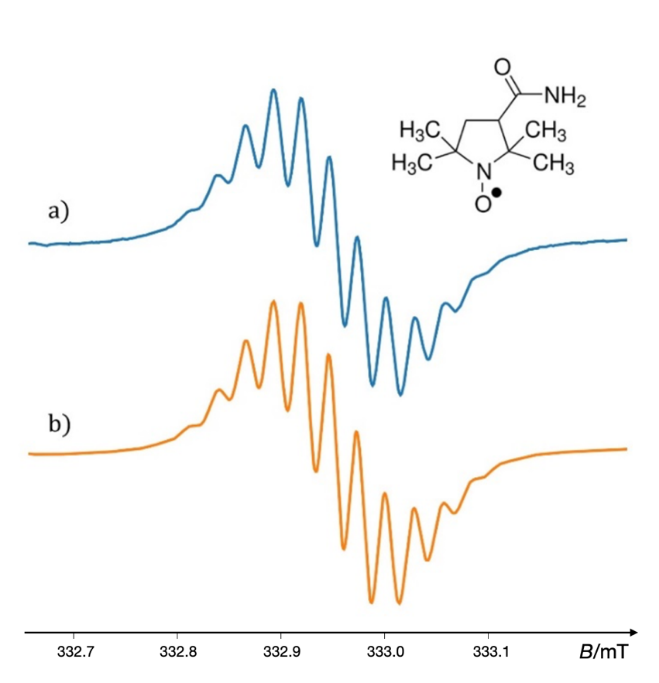
Fig. 18. Middle line of the EPR spectrum of the spin probe CTPO in fully deoxygenated toluene; a) experimental EPR spectrum; b) simulated spectrum in VisualEPR (177).
In addition to its application in organic chemistry, where the deoxygenation of samples is often required, EPR oximetry finds extensive use in in vivo medical research for monitoring oxygen levels in tissues, particularly in the context of diseases such as cancer and cardiovascular disorders, as well as in pharmaceutical studies assessing the efficacy of oxygen-dependent drugs. This versatility extends to its role in assessing tumor oxygenation, monitoring tissue oxygenation and blood flow, evaluating oxidative stress in the brain, and quantifying cellular respiration, including oxygen consumption by cells and mitochondria (190). Its applications extend to detecting ischemia in animal models, such as a saphenous artery-based flap in rats, where transcutaneous oximetry serves as a control. In one study, EPR oximetry and transcutaneous oximetry showed comparable results in measuring partial oxygen pressure under resting, ischemic, and post-tourniquet conditions (191). EPR spectroscopy's capability for non-invasive, real-time oxygen measurement offers a valuable tool for monitoring ischemic events and has potential clinical relevance, such as in surgical flap monitoring (192).
In terms of neurological applications, in vivo EPR has been successfully employed to measure partial oxygen pressure (pO2) in the brains of conscious rodents, utilizing lithium phthalocyanine crystals as oxygen-sensitive probes implanted in the cerebral cortex. This method enabled researchers to observe the cerebral pO2 changes induced by various anesthetics, including pentobarbital, ketamine/xylazine, and isoflurane, highlighting EPR spectroscopy's potential for studying the effects of neuroprotective agents in stroke and other pathological models (193). Similarly, the effects of vasodilators on muscle and tumor pO2 have been explored using a charcoal-based probe with high oxygen sensitivity. Their findings suggest that EPR oximetry may be applicable for pO2 measurement in peripheral tissues, particularly in patients with circulatory issues (194).
The development of low-frequency L-band EPR spectrometers and stable, biocompatible paramagnetic materials has further enhanced EPR oximetry's precision in vivo (195, 196). Oxygen-sensitive materials such as soluble radicals and particulate substances like charcoal, lithium phthalocyanine (LiPc), and Indian ink allow for extended monitoring of oxygen distribution in living tissues. Soluble radicals offer short-term measurements due to their reduction in biological systems, while particulate materials remain stable over prolonged periods, facilitating long-term studies (197, 198). Recent advancements have led to biocompatible oxygen sensors, including encapsulated paramagnetic particles, suitable for long-term in vivo studies without compromising oxygen sensitivity or causing toxicity (199, 200). Such sensors, based on biocompatible polymers, have shown stable in vivo performance for at least one month, demonstrating their potential for clinical trials (201).
The relationship between partial oxygen pressure (pO2) and line broadening in particulate materials shows either a linear correlation, as with LiPc, or a curvilinear trend with saturation at higher pO2 values, as seen with carbon-based materials. The sensitivity of various paramagnetic materials to pO2 changes, reflected by line width variations, varies across three orders of magnitude. However, this sensitivity alone does not directly indicate the relative utility of different materials for specific experiments. Considerations must also include the pO2 range in the system, the distribution of oxygen-sensitive spins, the time scale of the process, and the type of data required. Studies confirm that when properly inserted and equilibrated, paramagnetic materials do not significantly alter local pO2 measurements or physiological parameters, although it is essential to ensure minimal disturbance to physiology (192).
The stability of paramagnetic materials varies from short-term (approximately one week) for LiPc in muscle to potentially long-term (several years) for many carbon-based materials and LiPc in environments such as the spinal cord. Employing magnetic field gradients has enabled simultaneous spectral resolution at multiple sites, with successful applications in the heart (202), kidneys (203), liver (204), and brain (205).
EPR oximetry is particularly promising for tumor research due to its ability to measure and monitor pO2 levels, which significantly affect tumor response to radiation and chemotherapy. Although most studies have utilized 1 GHz frequencies, advancements with very low-frequency EPR spectrometers (250 ± 20 MHz) have measured oxygen concentrations in fibrosarcoma and assessed the impact of perfluorocarbon/carbogen on oxygen levels (206). In vivo EPR studies with oxygen-sensitive paramagnetic materials have demonstrated their feasibility in assessing pO2 in tumors over extended periods, tracking pO2 changes during tumor growth, and evaluating therapeutic effects. EPR imaging has also shown potential for mapping oxygen distribution in tumors, with preliminary studies providing images of oxygen tension and spatial resolution (207).
For brain studies, EPR oximetry enables the monitoring of pO2 in response to various physiological conditions and treatments. The technique has proven useful for evaluating brain pO2 under different breathing gases, anesthetics, and chronic hypoxia, offering insights into ischemia and reperfusion (204). In cardiac research, EPR oximetry has provided valuable data on oxygen dynamics and the effects of drugs and treatments on heart pO2. Despite challenges related to heart motion, both in situ and isolated heart studies have revealed critical insights into oxidative damage and therapeutic interventions (208). For skeletal muscle, EPR oximetry offers insights into muscle energetics, with studies utilizing both carbon-based materials and nitroxides. The technique's potential to integrate pO2 measurements with other metabolic indicators is promising (209). In liver research, EPR oximetry has effectively explored pO2 variations due to the organ’s complex blood supply and anatomy. The technique has revealed differences in pO2 between Kupffer cells and hepatocytes and has monitored the effects of toxins and ligation (210). EPR oximetry has also been applied to kidney research, overcoming technical challenges to provide pO2 measurements in both isolated and in vivo kidneys. Studies have highlighted the differential pO2 between the kidney cortex and medulla and identified the role of nitric oxide in mediating pO2 changes (203). Finally, EPR oximetry in the skin has potential clinical applications, especially for wound healing and peripheral vascular disease, due to the skin's accessibility for high-frequency EPR spectroscopy. Although current studies are limited, the technique's promise for assessing oxygen levels in skin and underlying tissues remains significant (192).
From a pharmaceutical perspective, EPR oximetry has proven valuable in evaluating oxygen changes after drug application. For instance, one study utilized EPR oximetry to examine the influence of different benzyl nicotinate (BN) formulations on skin oxygenation (211). They determined that lipid nanoparticles in hydrophilic gels provided the most efficient vehicle for BN delivery, outperforming liposomes and other formulations in maximizing skin oxygen levels. Similarly, another study explored the effects of liposomal size and composition on BN's efficacy, revealing that smaller liposomes induced faster increases in pO2 and more effective oxygen delivery to the skin (212). These findings underscore EPR oximetry's utility in tracking drug efficacy and oxygen dynamics over time.
In summary, EPR oximetry is a highly versatile technique for measuring oxygen concentration across a wide range of biological and chemical systems. Its ability to utilize oxygen-sensitive paramagnetic materials, such as soluble radicals and particulate substances, enables both short- and long-term oxygen monitoring. The method has shown significant potential in medical applications, particularly for assessing tissue oxygenation, tumor hypoxia, and ischemic conditions, while recent advancements in biocompatible materials have enhanced its clinical relevance. Moreover, EPR oximetry has proven effective in neurological and cardiac research, as well as in pharmaceutical studies, where it is used to evaluate drug-induced changes in oxygen levels. Its non-invasive, real-time capabilities make it a valuable tool in both experimental and clinical settings.
CONCLUSIONS
EPR spectroscopy emerges as a versatile and indispensable technique across various domains of pharmaceutical and biomedical research. Its exceptional sensitivity and specificity make it a pivotal tool for analyzing free radical scavenging properties, pharmacokinetics, and drug-radical interactions, providing profound insights into drug mechanisms, stability, and therapeutic efficacy. Specifically, the application of EPR spectroscopy in measuring radical scavenging, such as through the DPPH assay, has been crucial in evaluating the scavenging properties of pharmaceutical compounds, allowing for detailed characterization of their scavenging efficiency and long-term stability. EPR spectroscopy has also been instrumental in advancing our understanding of oxidative stress, drug-induced toxicities, and drug delivery systems, particularly through its ability to monitor molecular dynamics and interactions in real-time. In the field of metallodrug research, EPR spectroscopy has significantly contributed to elucidating the structural and redox properties of metallodrugs, optimizing their therapeutic potential, especially in oncology. Moreover, EPR oximetry’s ability to assess oxygen levels in biological systems has broadened its application in medical research, enabling precise monitoring of tissue oxygenation, ischemia, and tumor hypoxia. Although challenges such as sensitivity and penetration depth persist, ongoing technological advancements continue to enhance EPR spectroscopy’s utility in both in vitro and in vivo studies. Altogether, EPR spectroscopy remains a cornerstone in drug development, pharmacokinetics, drug delivery, and therapeutic optimization, offering unparalleled analytical power that bridges fundamental research and clinical applications.
Acknowledgments. – The authors would like to acknowledge financial support from the Croatian Science Foundation Installation grant UIP-2020-02-4857 LIGHT-N-RING. This work was also supported by the project FarmInova at the Faculty of Pharmacy and Biochemistry University of Zagreb (KK.01.1.1.02.0021) funded by the European Regional Development Fund.
Conflicts of interest. – The authors declare no conflict of interest.
Funding. – This research received no external funding.
Authors contributions. – Conceptualization, methodology, investigation, original draft preparation, writing, review and editing E.B.; original idea, writing, review and editing, Z.R.; analysis, visual representation, writing, review and editing, D.Š. All authors have read and agreed to the published version of the manuscript.