
Introduction
Polyurethane (PU) is a tailor-made class of polymers identified by carbamate links (─NHCOO). These polymers, especially thermoplastic PU (TPU), have demonstrated their role as well-known third-generation biomaterials (1, 2). A notable advantage is the use of the TPU matrix for composite biomaterials in bone tissue regeneration applications (3, 4). These are recognized by their high biocompatibility, bioresorption capacity, and physicochemical properties shaped by the internal structure of the polymer (5). An obvious objective for the synthesis of such composite materials is to enhance the acceptance and biocompatibility of biomaterials in the human body (6, 7). In such composites, bioactive inorganic materials have been frequently reported as fillers with a PU matrix to enhance proliferation, osteointegration, and osteoblast adhesion (8, 9). Yang et al. introduced the concept that natural bone structure can be mimicked by adding hydroxyapatite (HA) to the PU matrix. They initiated the application of porous aliphatic PU/HA composite scaffolds for bone tissue engineering (3).
HA [Ca10(PO4)6(OH)2] is an illustrious inorganic filler for PU composites due to its remarkable crystallographic and chemical resemblance to the mineral constituents of natural bone in bone tissue engineering (10, 11). HA contains a calcium/phosphorus ratio of 1.67, which is in close agreement with the composition of natural bone hard tissues. A composite of collagen with calcium and phosphorus minerals and HA constitutes bones. Moreover, the excellent biocompatibility, bioactivity, and osteoconductivity of HA enforced its application as a filler and implant material. It can stimulate bone regeneration and repair more efficiently due to its bioactive nature (12).
The characteristics of HA composites can be influenced by several surface modification techniques. Silane coupling agents can increase toughness and oleic acid treatment may improve mechanical properties. However, these modifications affected the bone repair property of modified HA composites (13). Modification of HA to improve biocompatibility in biological systems can also lead to adverse side effects. High temperatures and extended reaction time for modification of HA can cause the development of undesired calcium phosphate, which might be undesired for some applications (14).
A number of PU/HA composite scaffolds have been identified with promising potential for use in bone tissue engineering (15–17). Despite extensive research on the use of PU/HA composites for bone regeneration, there are still some issues to be addressed. One of the main concerns is the poor interaction of HA with the PU matrix because of the inorganic/organic interface. The extremity of this concern can be imagined by poor contact between the bioactive cells and filler as well as the weak adhesive forces, which could lead to implant loosening and ultimate failure. Thus, it is crucial to consider the proper interaction between the filler and matrix, which controls the ultimate structure and properties of composite biomaterials (18).
A momentous approach to improve the interaction of the filler with the polymer matrix is the surface modification of the filler (19). Therefore, a number of researchers have reported surface modification of HA with various other chemicals, such as silica, stearic acid, N-acetyl-L-cysteine, γ-benzyl-L-glutamate N-carboxyanhydride, triethanolamine (TEA), carbon nanotubes (CNTs), and di-isocyanates (17–23).
The surface modification of HA with isocyanates to prepare IHA dates back to 1995. Bos et al. reported a pretreatment of the HA particles with hexamethylene diisocyanate (HDI) before the addition to the crosslinked PU matrix. They claimed a significant improvement in the properties of PU/IHA composite materials. It was a very effective technique to improve the adhesion between the inorganic filler and the highly crosslinked polymer matrix (20). However, this study did not explore the biomedical application of PU/IHA composites. Xie et al. prepared shape memory PU/HA composite foams with isocyanate-modified HA nanoparticles. They proposed these shape memory foams for minimally invasive bone regeneration. They claimed no cytotoxicity and good in vitro biocompatibility of these prepared foams (4).
Through a groundbreaking investigation, researchers have strengthened the bond between hydroxyapatite (HA) and polyurethane (PU), going beyond simple physical mixing to form a strong chemical bond. PU, a flexible organic matrix, and HA, renowned for its inorganic strength, were combined not only at the surface but also at the molecular level. Diisocyanate is an intriguing chemical mediator that creates a strong connection between the flexibility of PU and the solidity of HA, opening up new possibilities for biomaterial composites.
In this innovative study, an attempt was made to develop an integrated interaction between HA and PU, transcending beyond mere physical mixing to a robust chemical fusion. HA, known for its inorganic attributes, and PU, a versatile organic matrix, were united not merely at the surface but at the molecular level. Diisocyanate, a potential chemical mediator to forge a durable link between HA’s solidity and PU’s flexibility, is promising a new horizon in biomaterial composites.
Henceforth, this study was designed to develop novel PU/IHA composites as scaffolds for bone tissue regeneration. First, HA was prepared by a hydrothermal method with high pressure and temperature. Second, surface modification of HA was carried out by 1,6-hexamethylene diisocyanate (HMDI). Then, two different levels (5 and 10 %) of IHA were incorporated into separately synthesized thermoplastic PUs by the solvent casting technique. FTIR and XRD analyses were used to monitor the whole synthesis process. The thermal stability and morphology of the PU/IHA composites were evaluated by TGA and SEM. Moreover, the cytocompatibility of pure PU and PU/IHA composites was investigated by a well-established MTT assay. The observations revealed an enhanced interaction between PU and IHA, which was constructive for cytocompatibility studies.
EXPERIMENAL
Materials
1,6-Hexamethylene diisocyanate (HMDI), isophorone diisocyanate (IPDI), poly( e-caprolactone) diol (PCL, Mr = 1000 and 2000), dibutylin dilurate (DBTDL) and 1,4-butanediol (BDO) were purchased from Sigma Aldrich Co., USA. Hydroquinone, tetrahydrofuran (THF), N,N-dimethylformamide (DMF), phosphoric acid, and calcium hydroxide of analytical grade were acquired from the Applied Chemistry Laboratory of the School of Chemistry, University of the Punjab, Lahore.
Synthesis of HA by the hydrothermal method
HA was synthesized by a hydrothermal method with a 1.67 mol ratio of Ca/P. A 0.5 mol L–1 solution of Ca(OH)2 was prepared by dissolving a calculated amount of base in distilled water. Similarly, a 0.3 mol L–1 solution of H3PO4 was prepared separately. Both of these solutions were mixed and stirred for two hours at ambient temperature. Then, the solution mixture was filled in a specially designed hydrothermal cell. The cell was made of good-quality steel with an inside lining of stable Teflon. This hydrothermal cell was placed in a closed oven at 180 °C for 24 hours. On the next day, the beautiful crystals of HA were separated by filtration, washed, dried in an oven, and ground into a fine powder (21).
Modification of HA to IHA
The surface modification of HA was carried out by HMDI. It was a chemical route of modification where –NCO of HMDI reacted with –OH of HA. In a practical protocol, a three-neck round bottom flask was charged with powdered HA (6 g). The inert environment was provided by the flow of N2 gas (100 mL min–1). Then, an appropriate amount of HMDI (6 mL) was added with constant stirring. Additionally, hydroquinone (0.2 g) and DBTDL (0.12 mL) were mixed in the reaction mixture. The temperature was maintained at 50 °C for the next 4 hours. Finally, the reaction mixture was removed from the flask, filtered, and rinsed three times with acetone and water. The wet white product was dried for 24 hours in an electric oven at 60 °C. The final product was a white powder of IHA (7).
Synthesis of PU-IHA composites
The primary monomers for PU synthesis were IPDI, PCL 1000 & 2000, and BDO. Two different compositions were optimized for the preparation of PU. Furthermore, four different PUR/IHA composites were prepared. The details of the sample codes, monomers used and composition of PU and respective composites are given in Table I. Moreover, a two-step method of synthesis was adopted for PU synthesis as reported earlier by our laboratory (8).
Table I. Codes of PU and PU-IHA composites, monomers, and percentage ratio of PU and IHA in composites
The composites of PU and IHA were prepared through a well-known solvent casting technique. A series of four different composites were developed and labeled PU1-IHA5, PU1-IHA10, PU2-IHA5, and PU2-IHA10. In a typical procedure, the PU solution was made by dissolving an accurately weighed amount of synthesized polymer in 30 mL of THF. The dissolution of PU was assisted by uninterrupted stirring and heating at 60 °C for one hour. On the other hand, a precisely weighed amount of prepared IHA was dispersed in another 30 mL of THF with stirring and heating at 60 °C for one hour. The solutions of PU and dispersion of IHA were combined and stirred continuously for the next three hours at 60 °C. A thick slurry was obtained at the end of the reaction. It was then poured onto a smooth Teflon plate and allowed to dry at room temperature. At the end of the procedure, a thin film was obtained (6).
Characterization of HA, IHA, PU, and PU/IHA composites
The structural analysis of PU1, PU2, and their respective composites was carried out by FTIR (Fourier Transform Infrared spectrophotometer). The particular model used was an FTIR Agilent Technologies Car 630, USA. The scan range was 650–4000 cm−1.
The thermal stability of composites with higher IHA loading (PU1-IHA10 and PU2-IHA10) was recorded by TGA (TGA-50, Shimadzu Corporation, Japan). The composites were heated from ambient temperature to 600 °C in a N2 environment. A heating rate of 10 °C min–1 was used. The flow rate of N2 gas was 50 mL min–1.
The crystal structure of IHA was elucidated by X-ray X'Pert Pro diffractometer Pan Analytical. Moreover, the XRD analysis helped to confirm the incorporation of IHA into the PU1 and PU2 composites. The XRD pattern was documented over a 2 θ range of 10–50°. A scan rate of 0.01° per minute was selected for this particular analysis. Scherrer’s formula ( Eq. 1) was used to calculate the crystallite size of hydrothermal HA and IHA as follows:
Dp = K λ/( B cos θ) ( 1)
In this formula, Dp is the average crystallite size (nm), and K is a constant dependent on crystallite shape. λ and B represent the X-ray wavelength used for analysis (Cu Kα average = 1.54178 Å) and FWHM (Full Width at Half Max) of the XRD peak, respectively. θ stands for the Bragg angle.
To make a comparison, the morphology of the synthesized PU1, PU2, and composites was investigated with TESCAN Vega LMU SEM. Different magnifications were used to capture clear images for each polymer and composite. The water absorption of PU1, PU2, and composites was calculated.
An SBF was prepared to study the biodegradability of polymers and composites. Samples (0.1 g), including polymer and composite, were dipped in 50 mL SBF for 30 days at ambient temperature (35 ± 3 °C). The weight loss of samples before and after was calculated.
The human colorectal HCT 116 cell line, with a key mutation in codon 13 of a gene significant to colorectal cancer, is a valuable model for studying tumor dynamics and drug responses. Exhibiting epithelial-like morphology, these cells are adaptable for various experimental designs, including monolayer and spheroid cultures. They are particularly useful for investigating the aggressive nature of tumors, cancer progression, and the underlying mechanisms of cancer cell behavior. Additionally, their high transfection efficiency makes them ideal for exploring signs and symptoms of initial-stage colon cancer. The cytocompatibility of PU1, PU2, and composites with the HCT 116 human colorectal cancer cell line ATCC®CCL-247™ (catalog no: 91091005-1VL Sigma Aldrich) was evaluated. A detailed procedure has already been reported by our laboratory (10). For in vitro cytotoxicity evaluation of prepared composites using an MTT bioassay. Adherent cells were washed, detached, and incubated. Different concentrations of actinomycete extracts were applied to the cells. After 24 hours, the MTT reagent was added, and the absorbance was measured. The inhibitory rate was calculated to assess the anticancer activities of the extracts. The entire experiment was performed in triplicate.
For statistical analysis, first, the means of the values were calculated to apply two-way ANOVA for multiple comparisons. In addition, a t-test was applied to evaluate the significant differences ( p < 0.05) among different samples.
Results and Discussion
Structural analysis by FTIR spectroscopy
FTIR spectroscopic analysis was performed to confirm the synthesis of HA, IHA, PU1, PU2, and all respective composites. The recorded spectra of these materials are shown in Fig. 1. Spectra of HA, modified IHA, PU1, and PU2 confirmed the synthesis and modification of HA with characteristic bands of –OH (stretching vibrations), CO32– (stretching and bending vibrations) and PO43– (stretching vibrations). The individual spectrum of HA exhibited a broad band of –OH stretching vibrations at 3183–3411 cm–1 and an intense band of PO43– stretching vibrations at 1022 cm–1. The stretching and bending vibrations of CO32– produced bands at 1401 and 870 cm–1, respectively (25, 26).
The spectrum of IHA showed a clearly modified pattern of bands. In the spectrum of IHA, the broad –OH band of HA was replaced by a narrow and intense band of –NH stretching vibration, which confirmed the development of a urethane link between –OH of HA and –NCO of HMDI. The –CH groups of IHA depicted symmetric and antisymmetric stretching vibrations at 2924 and 2855 cm–1. Moreover, the characteristic bands of –C=O and –NHCO of urethane links appeared at 1642 and 1553 cm–1, respectively. An important change to mention is the redshift in the characteristic stretching bands of PO43– and CO32– of HA after modification with HMDI. This may be ascribed to the development of intramolecular secondary interactions in the structure of IHA. A very short band of –NCO at 2357 cm–1 indicated the presence of few unreacted groups of HMDI, which would facilitate interactions among IHA particles and the PUR matrix. Hence, these characteristic changes in the spectrum of IHA substantiated the successful modification of hydrothermal HA.
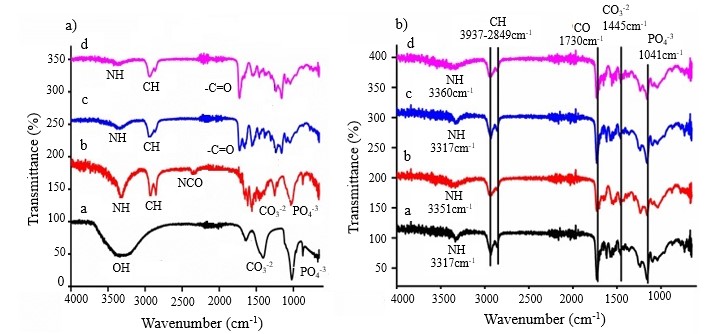
Fig. 1. FTIR spectra of: a) HA (a), IHA (b), PU1 (c) and PU2 (d); b) PU1-IHA5 (a), PU1-IHA10 (b), PU2-IHA5 (c) and PU2-IHA10 (d).
The spectra c and d in Fig. 1a) were produced by PU1 and PU2, respectively. The appearance of –NH stretching vibration bands at 3350 cm–1 for PU1 and at 3357 cm–1 for PU2 confirmed the development of urethane links. Furthermore, a band of –C=O stretching vibration at 1725 cm–1 supported the designed synthesis of these polymers (27).
The FTIR spectra of all four composites, i.e., PU1-IHA5, PU1-IHA10, PU2-IHA5, and PU2-IHA10, are shown in Fig. 1b). The characteristic bands of –NH (3317–3360 cm–1) and –C=O (1730 cm–1) of urethane links are clearly visible in these spectra. The other prominent bands of CO32– and PO43– of IHA were observed in the spectra at 1445 and 1041 cm–1, respectively. The presence of these bands confirmed the successful incorporation of IHA into PU matrices (28).
XRD analysis of HA, IHA, and PU-IHA composites
The XRD patterns of prepared HA and IHA are shown in Fig. 2a. The pattern of HA showed major peaks at 2 θ values of 25.90° (002), 31.91° (211), 32.10° (112), 39.37° (310), 43.14° (113), 47.53° (222) and 48.52° (320). The XRD pattern of IHA also showed a similar pattern, but the intensities of the peaks were compromised. This may be justified by the development of urethane links on the surface of the IHA. The hexagonal structure of HA and IHA was concluded by comparing the XRD data with JCPDS card 9–432 (29). The crystallite sizes of HA and IHA, as calculated by Sherr’s formula, were 11 and 20 Å, respectively. This increase in crystallite size reinforced the surface modification of IHA.
The XRD patterns of PU1-IHA5, PU1-IHA10, PU2-IHA5, and PU2-IHA10 are given in Fig. 2b. The composites revealed many characteristic peaks of IHA, including (hkl) 002, 210, 112, 211, 111, 310 and 200, which confirmed the incorporation of IHA in the PU matrices (22). Moreover, it was observed that the amorphous structure of PU was diminished by the incorporation of IHA and eventually vanished in the XRD patterns of PU2-IHA5 and PU2-IHA10.
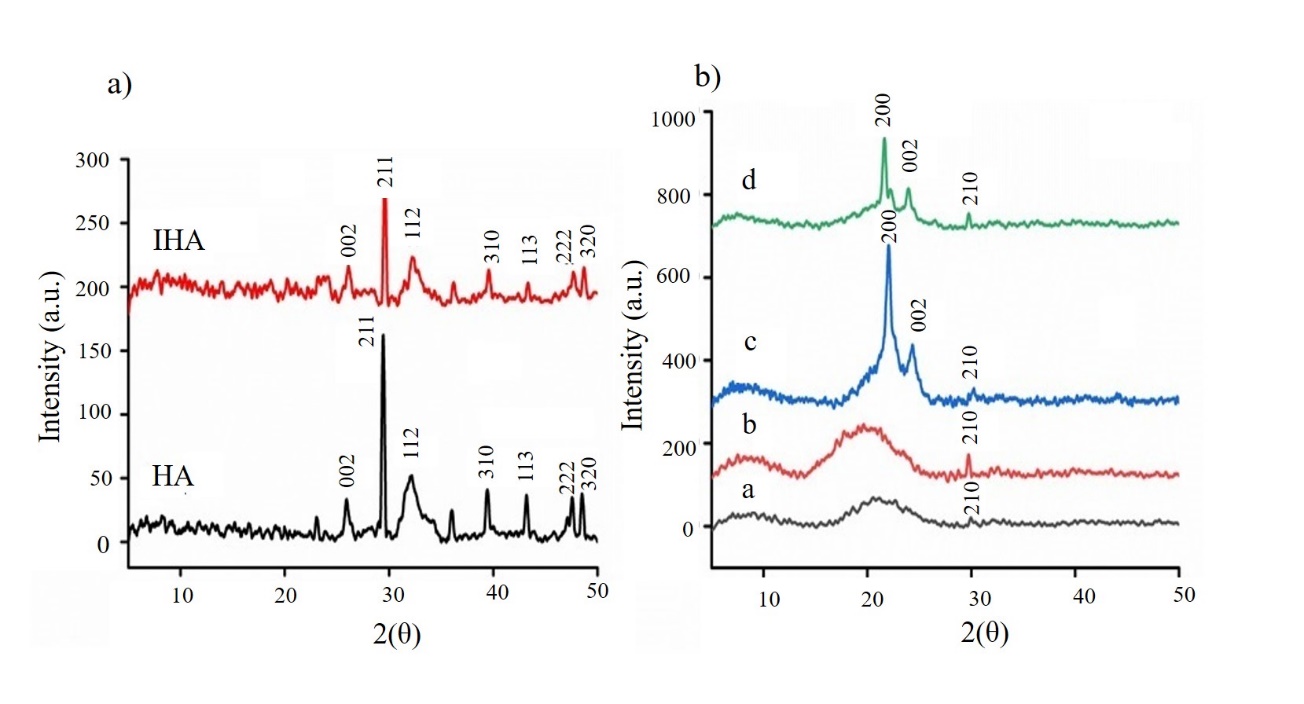
Fig. 2. a) XRD diffraction pattern of HA and IHA; b) X-ray diffraction pattern of composites: PU1-IHA5 (a), PU1-IHA10 (b), PU2-IHA5 (c) and PU2-IHA10 (d).
TGA of PU-IHA composites
Thermogravimetric analysis of a representative composite, i.e., PU2-IHA10, was performed. The loss in weight of the composite with heat was recorded and presented in Fig. 3 as TGA and DTG thermograms. The composite showed two stages of degradation. The first stage of degradation was associated with the breakage of the urethane links in the hard segment of the PU. The second stage of degradation appeared because of ester linkages in the soft segment of PU (7). The maximum degradation rates of PU2-IHA10 were found at 325 and 398 °C. These results revealed phase segregation in PU2-IHA10, which may be ascribed to the isocyanate modification of IHA. The presence of HMDI on the surface of HA enhanced the separation of soft and hard segments of the PU matrix. The residual weight obtained after complete degradation confirmed the presence of IHA in the composite (23).
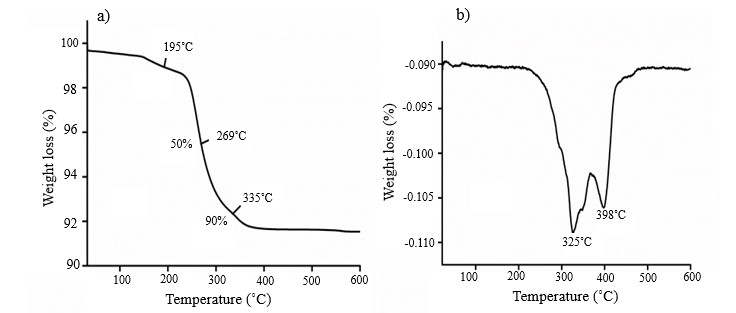
Fig. 3. a) Thermogram (TGA) of PU2-IHA10; b) derivatives of thermogram (DTG) of PU2-IHA10.
SEM of PU1, PU2 and PU-IHA composites
The morphology of the pure PU and its composites with IHA was recorded by SEM. The resultant micro images are displayed in Fig. 4. These images revealed a uniform interaction between modified IHA and PU matrices, as evident in Fig. 4, there is uniform morphology in all SEM images of PU1, PU2, PU-1HA5, PU2-1HA5 and PU-2HA10, no abruption or unoriented surface is seen. This may be ascribed to the surface modification of HA with HMDI, which helped to develop a few primary and mostly secondary interactions with PU matrices. Moreover, the incorporation of IHA markedly enhanced the roughness of the composites. This rough morphology of PU-IHA composites would be ideal for their intended application as biomaterials (8, 9).

Fig. 4. a) SEM images of: PU1 (a), PU2 (b), PU1-IHA5 (c), PU1-IHA10 (d), PU2-IHA5 (e) and PU2-IHA10 (f).
Cytotoxicity of PU-IHA composites
The cytotoxicity of materials including pure PU and composites was investigated through an MTT assay with antitumor cells. The quantitative results in terms of cell viability (%) for an incubation period of 28 h are shown in Fig. 5. In general, the cell viability of PU-IHA composites was significantly improved compared with that of pure PU matrices, which clearly indicated the constructive contribution of bioactive IHA. These findings were statistically supported by a two-way ANOVA. Furthermore, it was observed that a lower level of IHA, i.e., 5 %, in the composites resulted in better cell viability. Hence, it may be considered an optimized level for further studies (24).
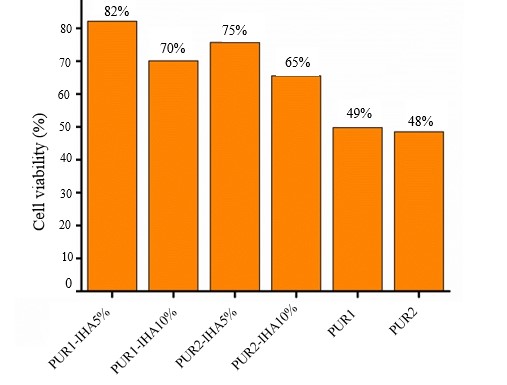
Fig. 5. Cell viability of PU1, PU2, and four composites. Data are represented as mean ± SD ( n = 3).
Water absorption and biodegradability of PU-IHA composites
The water uptake and biodegradability of the prepared polymers and composites were evaluated in simulated SBF. The results of the water absorption and biodegradation experiments were recorded as the weight gain and mass loss of the samples, respectively. Any change in the weight of the samples was calculated as a percentage and is presented in Fig. 6. PU2-IHA10 showed maximum mass loss (18.9 %). This may be justified by considering the long chains of the soft segment, i.e., PCL2000, and the higher content of IHA. Both of these factors may provide an opportunity for the ingress of water molecules into the composite, which accelerates the deterioration of the structure. In general, all of the composites demonstrated statistically significant ( p < 0.05) increased water uptake and better degradation in SBF than pure polymers. This result can be ascribed to the presence of IHA in the composites, which provided boosted hydrophilicity and ultimate weight loss. The higher molecular weight of the soft segment and higher content of IHA resulted in enhanced water uptake and weight loss. Most likely, the degradation of these composites followed the hydrolytic outbreak of ester linkages, which was accelerated by the hydrophilic nature of IHA.
The observed better results at 5 % IHA compared to 10 % IHA in terms of water uptake and biodegradability could be due to the optimal balance between hydrophilicity and structural integrity. At 5 % IHA, there is likely enough hydrophilic content to attract water molecules, which is necessary for biodegradation, without compromising the polymer’s structural stability. However, at 10 % IHA, the increased hydrophilicity might lead to excessive water absorption, causing a faster breakdown and loss of mechanical properties. This suggests that a moderate level of IHA incorporation can enhance biodegradability while maintaining the composite’s functional integrity.
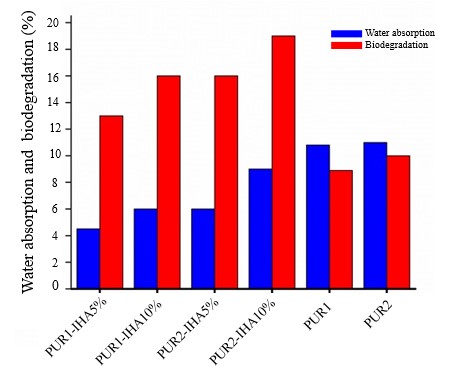
Fig. 6. Water absorption and biodegradation (%) in SBF of PU1, PU2, and PU-IHA composites. Data are represented as mean ± SD ( n = 3).
It is worth mentioning that the control unmodified HA was also studied, but the results were not encouraging enough to be reported. This part of the study affirmed the need for modification to improve the biocompatibility of HA.
Conclusions
The isocyanate-modified HA produced novel composites with PU that exhibited improved characteristics for biomedical applications. A simple solvent casting technique was quite supportive of composite preparation after the synthesis of individual moieties. A hexagonal crystalline structure of HA and IHA was confirmed by XRD. An increase in the crystallite size of HA from 11 to 20 Å for IHA was recorded with XRD. The FTIR spectra exhibited characteristic bands of urethane and HA, which confirmed the synthesis of the composites. Moreover, the XRD plots exposed an amorphous to semicrystalline nature of composites by incorporation of IHA. The composites displayed porous and granular morphologies, as captured by SEM. The TGA thermograms of the composites revealed a two-stage degradation of the composites. Furthermore, the cell viability of the composites was > 80 %, which was significantly higher than that of the pure polymers. These results endorse the great potential of synthesized PU-IHA composites for biomedical applications.
Data availability statement. – All the raw data in this research can be obtained from the corresponding authors upon reasonable request.
Acknowledgments. – The authors would like to extend their sincere appreciation to the Researchers Supporting Project Number (RSP2024R301), King Saud University, Riyadh, Saudi Arabia.
Conflicts of interest. – The authors declare no conflict of interest.
Authors contribution. – conceptualization, S.P-N.; methodology, M.S-N; analysis, M.U-N; writing, original draft preparation, M.S-N.; writing, review and editing, M.S-N, M.K-I and F.J-N. All authors have read and agreed to the published version of the manuscript.