
INTRODUCTION
Antimicrobial resistance is emerging as a major threat to global health (1). The World Health Organization (WHO) has published a list of bacteria for which new antibiotics are urgently needed. Of particular significance are ESKAPE pathogens, which are often multi-drug resistant: Klebsiella pneumoniae, Acinetobacter baumannii, Pseudomonas aeruginosa, Enterobacter spp ., Enterococcus faecium and Staphylococcus aureus (2). Most of the clinically used antibiotics target the biosynthesis of peptidoglycan, the main component of the bacterial cell wall, which is present in both Gram-positive and Gram-negative bacteria (3). The bacterial cell wall gives bacteria their shape and rigidity, maintains proper osmotic pressure, and allows them to survive in a hypotonic environment by protecting them from lysis (4). Bacterial cell wall biosynthesis occurs in three phases, taking place in the cytoplasm, the inner membrane, and finally in the periplasm. Several successive steps in the cytoplasm lead to the formation of the monomeric building block precursor UDP- N-acetylmuramic acid pentapeptide, which, upon association with N-acetylglucosamine, leads to the generation of lipid II, the main peptidoglycan building block. After transport across the membrane, lipid II is incorporated into the peptidoglycan through the action of penicillin-binding proteins (PBPs) and SEDS (shape, elongation, division, and sporulation family proteins (5). PBPs catalyze two sequential steps, namely, the polymerization of glycan strands (transglycosylation), and stem peptide cross-linking (transpeptidation), forming a three-dimensional network ( Fig. 1) (6–10). Inhibition or deregulation of peptidoglycan biosynthesis often leads to impaired cell growth, shape defects, cell lysis, and death.

Fig.1. The versatile roles of penicillin-binding proteins (PBPs) in the biosynthesis of peptidoglycan.
Since 1941, when penicillin was first used in humans, β-lactam antibiotics have remained the largest and most important structural class of antibiotics worldwide (11–13). β-lactams ( i.e., penicillins, cephalosporins, carbapenems, and monobactams), whose core structure mimics the terminal D-Ala- D-Ala unit of the native enzyme substrate, act as irreversible inhibitors of the cross-linking reactions between the nascent peptidoglycan chains during the final stages of peptidoglycan biosynthesis (14) via acylation of catalytic serine residues in the active sites of transpeptidase and carboxypeptidases, including PBPs. The hydroxyl group of a serine residue reacts with the lactam carbonyl resulting in the opening of the ring. The resulting acyl-enzyme complex is stable, and its hydrolysis proceeds very slowly (15, 16). Monocyclic β-lactams are four-membered cyclic amides with an oxo group at a second position of the ring and various substitutions at the amide nitrogen (N1), at the C3 carbon adjacent to the carbonyl group, and at the C4 carbon adjacent to the nitrogen (17). Based on the N1 substituents, they are divided into several classes (monobactams, monosulfactams, oxamazines, thiamazines, monocarbams, and nocardins), which also determine their chemical reactivity (18). The first monocyclic β-lactam, nocardicin A, was discovered in 1976 in the bacterium Nocardia uniformis. Subsequently, sulfazecin and isosulfazecin were isolated from Pseudomonas strains (19). The first synthetic monocyclic β-lactam with antibacterial activity in clinical use was aztreonam ( Fig. 2) (20). Recently, ancremonam ( Fig. 2), which has shown potent activity against Enterobacteriaceae and is also stable against serine β-lactamases, has completed the second phase of clinical trials (21–23). The second promising new monocyclic β-lactam is AIC499 developed by AiCuris (Fig. 2). Monocyclic β-lactams mainly target PBP3 and have a limited spectrum of activity against Gram-negative bacteria, including Pseudomonas aeruginosa, and good stability against β-lactamases. Particularly important is their stability against metallo-β-lactamases (24–29). Although β-lactams are relatively non-toxic, as already shown by Fleming, there is the possibility of a rare idiosyncratic immune reaction to penicillins; however, there is no cross-reactivity to aztreonam (30).
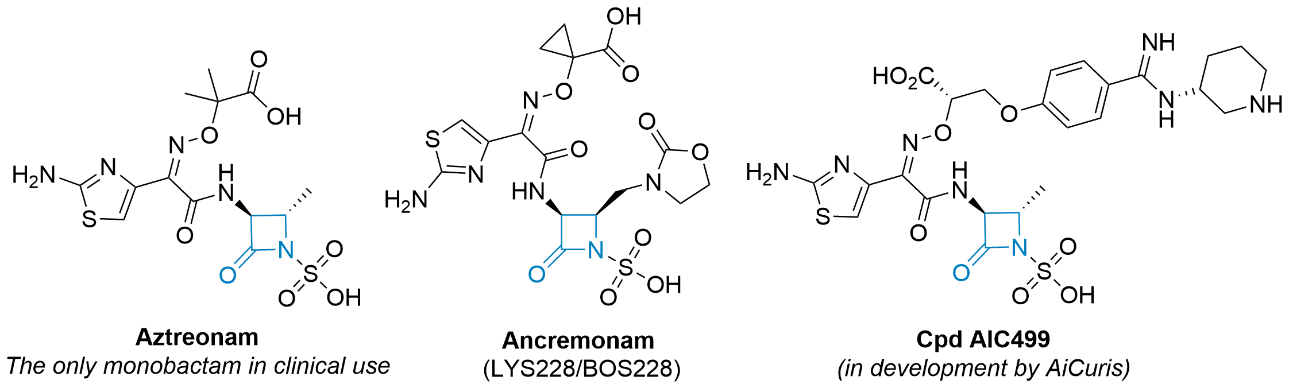
Fig. 2. Selected examples of monocyclic β-lactam compounds (β-lactam ring is highlighted in blue).
Amongst several bacterial resistance mechanisms, such as, e.g., PBP mutations or the expression or alterations of porins, the production of β-lactam-hydrolyzing enzymes ( i.e., β-lactamases) remains the clinically most important mode of resistance to this structural class (31–34). Pharmaceutical industry research in this area is inadequate because new antibacterial agents are likely to be classified as "reserve" antibiotics in the WHO’s AWaRe classification (35), making them even less economically viable; solutions are being sought in public-private partnerships and government programs such as IMI, CARB-X and the AMR Action Fund (36, 37)
Monocyclic β-lactams, which can be assembled synthetically, are active against some ESKAPE pathogens and currently appear to resist metallo-β-lactamase-mediated hydrolysis (34). Herein, we present the synthesis and biological evaluation of a focused library of novel 3-amido-4-substituted monocyclic β-lactams. Analogues bearing various aliphatic, aromatic, and heteroaromatic substituents at the C4 position, and amides with various carboxylic acids from known antibiotics at the C3 amino group were prepared. We also incorporated various substitutions at N1 in the lactam ring that affect the reactivity of the ring. The compounds were evaluated for their inhibition of PBP1b from the human pathogen S. pneumoniae (38), and for their antibacterial activity against a panel of Gram-positive and Gram-negative bacteria. To further characterize the series, the compounds were evaluated for their aqueous stability, initial chemical reactivity with the cysteine surrogate, and activity against other serine and cysteine hydrolases.
EXPERIMENTAL
General chemistry
Chemicals and solvents were from commercial sources (Sigma Aldrich, USA; Acros Organics, Belgium; TCI, Japan; Fluorochem, UK and Apollo Scientific, UK) and were used as supplies. Dry tetrahydrofuran was prepared by distillation from a mixture of sodium and benzophenone.
Reactions were monitored by thin-layer chromatography (TLC) on 0.25-mm silica gel 60F254 plates (Merck KGaA, Germany). Flash column chromatography was performed on silica gel 60 (Merck KGaA, particle size 0.040–0.063 mm) using the indicated solvents in each individual synthetic step. Yields are given for purified products.
1H and13C NMR spectra were recorded on a Bruker AVANCE III 400 (USA) spectrometer at 295 K in commercially available deuterated solvents (as indicated) with TMS as the internal standard. Chemical shifts ( δ) are reported in parts per million (ppm) downfield from TMS. Coupling constants ( J) are given in Hertz (Hz), and splitting patterns are given as follows: s, singlet; br s, broad singlet; d, doublet; dd, doublet of doublets; t, triplet; dt, doublet of triplets; m, multiplet. Mass spectra were recorded using an ADVION Expression CMSL mass spectrometer (Advion Inc., USA) with ESI ionization. High-resolution mass spectra were obtained with the ExactiveTM Plus Orbitrap mass spectrometer with ESI ionization (Thermo Fisher Scientific Inc., USA).
Full experimental procedures (including analytical data) are available in the Supplementary materials.
Expression and purification of S. pneumoniae PBP1b
A vector expressing PBP1b from S. pneumoniae (pGEX-GST-PBP1b) was used to transform chemically competent Escherichia coli NiCo21(DE3) (New England Biolabs, USA), as previously described (39, 40). Cells were cultured at 37 °C and shaken at 250 rpm in LB broth containing 100 µg mL–1 ampicillin until an OD600 ≅ 1 was achieved. Expression was induced by the addition of 1 mmol L–1 IPTG and cultured at 16 °C for an additional 20 hours. Cells were harvested by centrifugation (10 min, 3000× g, 4 °C), and cell pellets were stored at –80 °C until purification. Cell pellet was resuspended in buffer A (50 mmol L–1 Tris × HCl, 200 mmol L–1 NaCl, 1 mmol L–1 EDTA, 1 mmol L–1 DTT, pH 8.0) and lysed on ice by sonication. Cell debris was removed by centrifugation for 30 min (16,000× g, 4 °C, repeated twice). The cleared lysate was loaded onto two interconnected 1-mL GSTrap HP columns (Cytiva, USA), which were pre-equilibrated with buffer A. The column was washed with buffer A and the protein was eluted with buffer B (50 mmol L–1 Tris, 200 mmol L–1 NaCl, 1 mmol L–1 EDTA, pH 8.0, 1 mmol L–1 DTT, 10 mmol L–1 reduced glutathione). Eluted PBP1b was transferred to buffer C (50 mmol L–1 HEPES, 100 mmol L–1 NaCl, 1 mmol L–1 EDTA, 10 % glycerol, pH 7.0) by buffer exchange. The protein was concentrated with a 50-kDa molecular mass cut-off filter (Amicon® Ultra-4 Centrifugal Filter Unit, Merck KGaA), aliquoted, frozen in liquid nitrogen, and stored at –80 °C. Protein purity was assessed using SDS-PAGE, and the concentration was determined fluorometrically using Invitrogen Qubit (Thermo Fisher).
PBP1b inhibition assay using Ellman reagent
Inhibition of S. pneumoniae PBP1b was measured spectrophotometrically by measuring the formation of 2-nitro-5-thiobenzoate anion (TNB2–) during the reaction; residual activities were determined based on the ability of a potential inhibitor to prevent hydrolysis of the substrate analog thioester 2-{[(benzoyl-D-alanyl)-thio]-acetic acid} as described previously (40). PBP1b (0.4 µmol L–1) was incubated with the compound (final concentration 100 µmol L–1) in 10 mmol L–1 sodium phosphate buffer (pH 7.0) in the presence of 100 mmol L–1 D-alanine, 0.01 mg mL–1 BSA, and 0.01 % Triton X-100 for 60 minutes at 25 °C. After preincubation, 5,5'-dithiobis-(2-nitrobenzoic acid) (DTNB, Ellman reagent) and thioester were added to initiate the reaction and reach a final concentration of 1 and 5 mmol L–1, resp. The final volume of the reaction mixture was 150 µL. Triton X-100 was added to minimize the detection of false positives (promiscuous inhibitors). The initial rate of thioester hydrolysis was determined by measuring absorbance at 412 nm for 30 min using a 96-well microtiter plate using a BioTek Synergy H4 Hybrid microplate reader (BioTek Instruments, USA). The same assay was performed in the absence of the inhibitor (1 %, V/V, DMSO).
Aztreonam, which completely inhibits PBP1b [RA (at 500 µmol L–1) = 1.4 ± 0.1 %; IC50 (60 min pre-incubation) = 1.2 ± 0.1 µmol L–1], was used as a positive control. All experiments were performed in triplicate. The ratio of the reaction rate with inhibitor ( vi) to the reaction rate without it ( vo), gives the residual activity ( RA) expressed as a percentage:
RA = [( vi – b)/( vo – b)] × 100
where b is the blank value for the initial rate of spontaneous hydrolysis of the thioester in the presence of the inhibitor and in the absence of PBP1b. IC50 values were determined by measuring the reaction rates at seven different inhibitor concentrations using a non-linear regression (four-parameter model) applied in GraphPad Prism 9.0.2 (GraphPad Inc, USA).
PBP1b inhibition assay with BOCILLIN FL
A complementary assay to measure the inhibition of S. pneumoniae PBP1b with BOCILLIN FL was also used (41). Fluorescence anisotropy was measured using 60 nmol L–1 purified PBP1b, 30 nmol L–1 BOCILLIN FL in 100 mmol L–1 sodium phosphate buffer pH 7.0 containing 0.01 % Triton X-100 to reduce promiscuous inhibitor detection and protein binding to the plate. The assay was performed in triplicate in a volume of 50 μL in black flat-bottom, 384-well microplates at 30 °C. The change in fluorescence anisotropy ( FA) was measured using a Biotek Synergy H4 Hybrid microplate reader with polarizing filters at excitation λ = 482 nm and emission λ = 530 nm and calculated using the following equation:
FA = ( Fpara – Fperp)/( Fpara + 2 Fperp)
where Fpara is the fluorescence intensity parallel to the excitation plane and Fperp is the fluorescence intensity perpendicular to the excitation plane. Residual activities were determined by preincubating the test compound (100 µmol L-1) and the protein for 1 h at 30 °C before initiating the reaction by adding BOCILLIN FL. To determine the residual activity, the change in FA after 30 minutes was compared to the uninhibited (1 %, V/ V, DMSO) control.
Inhibition of cholinesterases
The inhibitory potency of the compounds toward hBChE and hAChE was determined by the Ellman method according to the procedure described previously (42). Briefly, stock solutions of the compounds in DMSO containing DTNB and ChEs (final concentrations: 370 μmol L–1 DTNB, 1 nmol L–1 or 50 pmol L–1 recombinant hBChE, or recombinant hAChE, resp.) were incubated in 0.1 mol L–1 sodium phosphate pH 8.0 for 60 min at 20 °C. Reactions were started by adding the substrate (final concentration equal to 500 µmol L–1 butyrylthiocholine iodide or acetylthiocholine iodide for hBChE and hAChE, resp.). The final DMSO concentration was always 1 % ( V/V). The increase in absorbance at 412 nm was monitored for 2 minutes using a 96-well microplate reader (BioTek Synergy H4, BioTek). The initial velocities in the presence ( vi) and absence ( vo) of the test compounds were calculated. The inhibitory effect was expressed as residual activity, corresponding to RA (%) = ( vi/ vo) × 100.
Inhibition of 3CLpro
The enzymatic activity of 3CLpro was measured by a kinetic assay using the fluorogenic FRET substrate DABCYL-KTSAVLQSGFRKME-EDANS (CPC Scientific, USA). Experiments were performed in an assay buffer containing 50 mmol L–1 Tris-HCl pH 7.3, 1 mmol L–1 EDTA, and 0.05 % Triton X-114. Briefly, compounds were pre-incubated at a concentration of 100 µmol L–1 with 3CLpro for 30 min at 30 °C. The reaction was started by the addition of substrate, and the increase in fluorescence intensity was measured using a Synergy H4 microplate reader (BioTek) at λex = 360/40 nm and λem = 440/40 nm. The final concentrations were as follows: compound, 100 µmol L–1; substrate, 20 µmol L–1; 3CLpro, 50 nmol L–1; DMSO, 10 % ( V/ V). For the control experiments, the compound was replaced by DMSO. For the determination of b (blank), the enzyme was replaced by Tris-HCl buffer. The initial velocities ( v) were calculated from the linear trends obtained, each measurement being performed in duplicate. The inhibitory potency was expressed as the residual activity ( RA):
RA = ( vi – b)/( vo – b)
where vi is the velocity in the presence of the test compound, and v0 is the control velocity in the presence of DMSO. To check for spectral interference, absorbance at the excitation and emission wavelengths and autofluorescence were determined for the active compounds in the buffer solution. Boceprevir and carmofur (at 100 µmol L–1 concentration in the assay) were used as positive controls with a residual activity of 4.8 ± 0.5 and 34.8 ± 2.8 %, resp.
Antimicrobial susceptibility testing
Minimum inhibitory concentrations ( MICs) were determined by the broth microdilution method in 96-well U plates according to CLSI guidelines and European Committee on Antimicrobial Susceptibility Testing recommendations (44, 45). Suspensions of specific bacterial strains ( S. aureus ATCC 29213, E. coli ATCC 25922, K. pneumoniae RDK 070A (ATCC 51503), P. aeruginosa RDK 184 (ATCC 15442), E. faecalis ATCC 29212, E. coli N43 (CGSC no. 5583) and E. coli D22) (CGSC no. 5163) corresponding to the 0.5-McFarland turbidity standard were diluted with cation-adjusted Mueller-Hinton broth with TES to yield an end inoculum of 5 × 105 CFU mL–1 for the assay. The compounds, dissolved in DMSO, and the bacterial inoculum were mixed and incubated at 35 °C for 18–24 hours. The MIC values were determined by visual inspection as the lowest dilution of the compounds that did not exhibit turbidity. Tetracycline was used as a positive control on each test plate. All experiments were performed in duplicate.
Thiol reactivity assay
The thiol reactivity of the compounds with DTNB was determined according to a previously published protocol (46, 47). TNB2– anion was prepared in situ from DTNB and tris(2-carboxyethyl)phosphine hydrochloride (TCEP). The reaction was performed in a 96-well microplate at 37 °C with a final volume of 300 μL buffer (20 mmol L–1 sodium phosphate buffer, 150 mmol L–1 NaCl, pH 7.4) containing 100 μmol L–1 compound, 100 μmol L–1 TCEP, 25 μmol L–1 DTNB, and 1 % ( V/V) DMSO. The plate was incubated at 37 °C in a plate reader (Synergy H4, BioTek), and absorbance was recorded at 412 nm every 5 minutes for 12 hours. Each compound was measured in duplicate, and a parallel experiment was performed without DTNB to determine the background absorbance of the compound, which was then subtracted from each measurement. The second-order rate constant ( k) was calculated using the equation:
where [A0] and [B0] are the initial concentrations of the compound and TNB2–, resp., and [A] and [B] represent the remaining concentrations of TNB2– and the compound as a function of time. Iodoacetamide was used as a positive control.
Buffer stability assay
The stability of the compounds in phosphate buffer was measured at different pH values (pH 7.0 and pH 8.0) as described previously (56). Stock solutions of the investigated compounds were prepared in DMSO. The final concentration of each studied compound in 10 mmol L–1 phosphate buffer was 50 µmmol L–1, with 5 % ( V/V) DMSO. The reaction was carried out in a 96-well microplate. The plate was incubated at 37 °C in a plate reader (Synergy H4, BioTek) for 120–240 min. Absorbance values were measured in sweep mode after 0, 15, 30, 60, 120, 180, and 240 min using a discontinuous kinetic procedure in Gen5 software (BioTek). The time required to read the entire 96-well plate was 3 min. To determine the baseline, the compound solution was replaced with pure DMSO and subtracted from each reading. The relative absorbance difference between the first time point and 240 min at the most responsive wavelength was calculated. If the relative absorbance difference for the compound in the buffer was below 0.1, between 0.1–0.2, and above 0.2, the compound was classified as stable, intermediate, and unstable, resp.
RESULTS AND DISCUSSION
Chemical synthesis
In a previous publication, we described our initial development and optimization efforts to prepare a range of desired 3-amino-4-substituted monocyclic ß-lactam intermediates using Staudinger cycloaddition (48). In our quest to obtain biologically active compounds, further derivatization at the C3 position and the activation of the N1 position were envisaged. Primarily, N-sulfonation (18) was used as the most common activation method to obtain N1-activated monocyclic β-lactam derivatives, as summarized in Scheme 1.
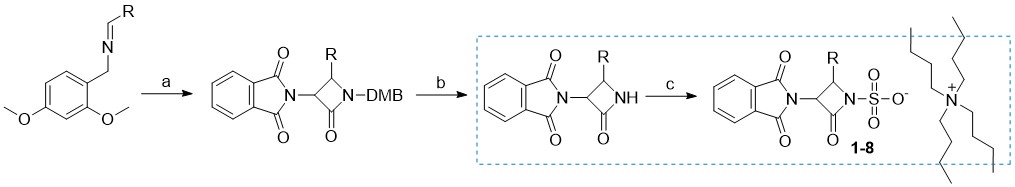
Scheme 1
Reagents and conditions: a) 2-(1,3-dioxoisoindolin-2-yl)acetyl chloride, Et3N, toluene, 70 °C; b) (NH4)2[Ce(NO3)6, CH3CN/H2O, –10 °C; c) SO3×DMF, DMF, K2HPO4–, nBu4NHSO4.
Briefly, C-3 pthalimido N1–SO3–TBA+ monocyclic β-lactam analogs 1–8 (detailed structures shown in Supplementary materials, Table SI) were prepared from corresponding 2,4-dimethoxybenzyl (DMB) N1 protected β-lactams by sulfonation with an excess of SO3×DMF complex in anhydrous dimethylformamide at room temperature (49). In some cases, the sulfonation reaction took several days, despite a large excess of reagent added. Once the reaction was complete as monitored by TLC analysis, the sulfonated intermediates were isolated in the form of tetrabutylammonium (TBA) salts. Note that in the case of free amino analog 5, an extra synthetic manipulation was necessary to obtain the compound (catalytic hydrogenation of nitro compound 4; see S upplementary materials for more details). We then turned our attention to the preparation of novel monocyclic β-lactams with diverse substituents at the C4 position and different aminothiazolemethoxime (ATMO) side chains. We have used two different approaches, starting from intermediates with either Boc or Fmoc protecting groups ( Scheme 2 ). To obtain the intermediates with the desired aromatic 4-substitutions on the monocyclic β-lactam core, appropriate imines were first prepared by condensation of dimethoxybenzylamine with aromatic aldehydes in dichloromethane at room temperature, using an excess of anhydrous sodium sulfate as a drying agent ( 9–12). Subsequent Staudinger cycloaddition of imines with activated N-phthaloylglycine ( i.e., in its acyl chloride form) afforded monocyclic β-lactams 13–16 which were easily isolated in high purity by precipitation or flash column chromatography. Since deprotection of the phthalimido (Phth) group generally requires relatively harsh conditions, we opted to remove it in the next step and replace it with carbamate-protecting groups, which can be more easily removed. To this end, the Phth protecting group was cleaved with methylhydrazine and the resultant C3 free amine was directly protected with tert-butyloxycarbonyl (Boc, compounds 17–18) or fluorenylmethoxycarbonyl (Fmoc, compounds 19–20) protecting group. The resultant monocyclic β-lactams were then subjected to the oxidative cleavage of the N1–DMB moiety with cerium ammonium nitrate under mild conditions, to afford the desired N1–H building blocks 21–24. For the Boc-protected intermediates 21 and 22, the Boc-protecting group was removed in high yields by trifluoroacetic acid with triethylsilane used as a scavenging agent. With the free C3 aminoazetidin-2-ones in hand, we initially tried to couple the selected ATMO side-chains with the aid of the common amide coupling reagents, such as, e.g., HATU or TBTU. Because these efforts were unproductive we turned our attention to the coupling reactions between N-hydroxysuccinimide (NHS) ester-modified ATMO side chains and amines under basic conditions.
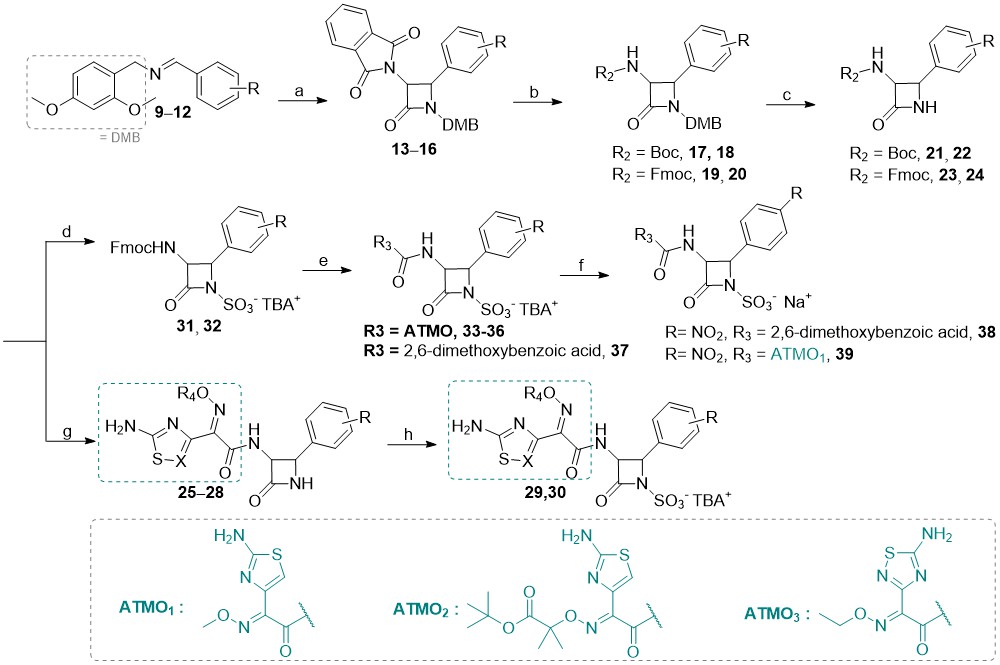
Scheme 2
Reagents and conditions: a) 2-(1,3-dioxoisoindolin-2-yl)acetyl chloride, Et3N, toluene, 70 °C; b) CH3NHNH2, CH2Cl2, rt, then fluorenylmethyloxycarbonyl chloride, N,N-diisopropylethylamine, CH3CN, 0 °C or Boc2O, Et3N, CH3OH, rt; c) (NH4)2[Ce(NO3)6, CH3CN/H2O, –10 °C; d) SO3×DMF, DMF, then K2HPO4–, nBu4NHSO4; e) Et3N, CH3CN, rt, then NHS-activated carboxylic acid side-chain, DMF, 70 °C; f) Dowex® (50WX8, Na+ form), THF/H2O, rt; g) trifluoroacetic acid, triethylsilane, CH2Cl2, 0 °C, then NHS-activated carboxylic acid sidechain, DMF, 70 °C; h) SO3×DMF, DMF, then K2HPO4-, nBu4NHSO4; i) Dowex® (50WX8, Na+ form), THF/H2O, rt.
Surprisingly, when we subjected crude amines 21 and 22 to conditions featuring NHS-activated ATMOs in DMF at 70 °C, a diverse set of amide compounds 25–28 was obtained. Next, the treatment with an excess of SO3-DMF complex furnished the desired N-sulfonated β-lactams 29 and 30 as TBA salts. In the case of the Fmoc-protected intermediates 23–24, we first formed N1–SO3–TBA+ monocyclic β-lactams 31–32, and subsequently removed the Fmoc group under basic conditions. Coupling of crude amines with NHS-activated ATMO side chains yielded analogs 33–37. Additionally, the less hydrophilic 2,6-dimethoxyphenylacetamido side-chain of methicillin was also incorporated in analog 38. For all TBA salts, the last step we envisaged was a cation exchange (to obtain the corresponding sodium salts) using Dowex® resin (50WX8, Na+ form). Unfortunately, except for the methicillin analog 38 and compound 39, the conversions of N1–SO3–TBA+ β-lactams to the corresponding Na+ form were unsuccessful, as the compounds likely remained bound to the Dowex® cation exchange resin, and could not be successfully washed off. Hence, this step must be optimized in future synthetic efforts. Note that under the conditions used the products of the Staudinger [2+2]-cycloaddition reaction was isolated as cis-isomers since electron-withdrawing groups on the imine facilitate a direct ring closure (in turn, this also leads to better yields); the cis-configuration of the newly synthesized monocyclic β-lactams was deduced from the corresponding1H NMR coupling constants of the β-lactam ring hydrogens H3 and H4 (see sample spectra in Supporting material); for cis-β-lactams J3–4 ~ 5–6 Hz and for trans-β-lactams J3–4 ~ 2 Hz, consistent with the literature data (15).
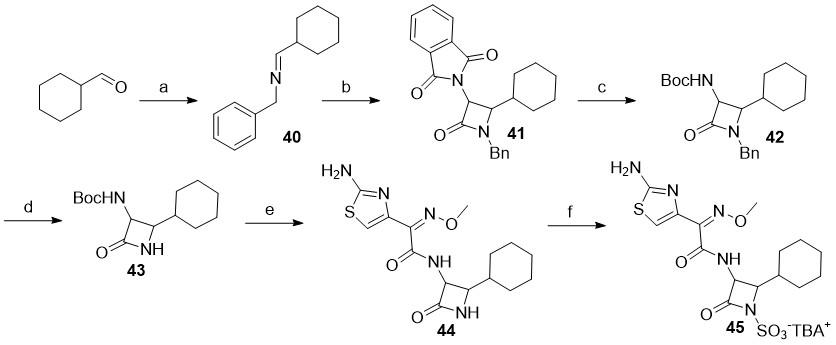
Scheme 3
Reagents and conditions: a) benzylamine, Na2SO4, CH2Cl2, rt; b) 2-(1,3-dioxoisoindolin-2-yl)acetyl chloride, Et3N, toluene, 70 °C; c) CH3NHNH2, CH2Cl2, rt, then Boc2O, Et3N, CH3OH, rt; d) SD Super Fine™ (sodium 25 %, m/ m, dispersion in mineral oil), 15-crown-5, i-propyl alcohol, THF, 0 °C; e) trifluoroacetic acid, triethylsilane, CH2Cl2, 0 °C, then NHS-activated carboxylic acid sidechain, DMF, 70 °C; f) SO3×DMF, DMF, then K2HPO4-, nBu4NHSO4.
In the case of monocyclic β-lactams with aliphatic substituents at the C4 position ( i.e., Scheme 3), the synthesis was more challenging. Although we attempted to prepare several different analogs with aliphatic substituents ( e.g., cyclohexane, cyclopropyl, i-propyl …), only the C4 cyclohexane analog 45 was successfully synthesized. Whilst the synthetic conditions used were largely similar to the ones described earlier, we were careful to execute: ( i) the imine condensation reactions on ice and use them directly without evaporation of the solvent (due to increased reactivity of aliphatic aldehydes), and ( ii) the cycloaddition reaction at room temperature to afford a monocyclic β-lactam. Since the deprotection of the N1-DMB protecting group in analogs bearing aliphatic side-chains at C4-position was unproductive, we elected to utilize the benzyl (Bn) group for N1 protection instead; the key N1-benzyl intermediate 42 was therefore synthesized in three steps starting from commercially available cyclohexanecarbaldehyde. Ammonia-free Birch reduction of 42 gave the desired intermediate 43 in excellent yield and with almost no by-products. Following the Boc cleavage, coupling with NHS-activated ATMO side-chain and subsequent N1-sulfonation of 44, the N-sulfonated monocyclic β-lactam 45 was isolated as a TBA salt.
Biological and reactivity evaluation of monocyclic β-lactams
Compounds of interest were first tested on PBP1b in a biochemical assay on microtiter plates using a thioester analog of the substrate (S2d) and detection with Ellman reagent at λABS = 412 nm (40). The first series of N1-activated monobactams we tested were TBA salts of C3–phthalimido N-sulfonated β-lactams 1–8 (Supplementary material, Table SI). Expectedly, these compounds did not show any inhibition of PBP1b. Then, the second series featuring fully functionalized novel analogs ( Table I) was evaluated in the same assay. Compound 38 with a 2,6-dimethoxybenzoic acid ( i.e., ‘methicillin mimic’) substitution at the C3 position was also not active. However, other monocyclic β-lactams ( e.g., 29–30, 33–36, and 45) with various ATMOs attached at the C3 position showed modest PBP1b inhibitory activity. Overall, the IC50 values measured after one hour of pre-incubation with the enzyme showed 5- to 15-fold less potential than the positive control ( i.e., aztreonam, IC50 = 1.3 ± 0.4 μmol L–1) for compounds 30, 34 and 45 ( IC50 values were 6.4, 14.8 and 18.7 μmol L–1, resp.), while the values for the remaining analogs were about 60-fold higher. The less soluble cyano derivative 33 was also not very active at a concentration of 100 μmol L–1, exhibiting only ~50 % residual activity in both PBP1b inhibition assays.
Table I. Residual activities of novel cis-C3/C4 functionalized N-sulfonated β-lactams isolated as TBA+ saltsa
a Residual activities were determined at a concentration of 100 µmol L –1 of the tested compounds in the assay after 60 min of pre-incubation with the enzyme. ATMO1 – 2-(2-aminothiazol-4-yl)-2-(methoxyimino)acetate, ATMO2 – 2-(2-aminothiazol-4-yl)-2-(((1-( tert-butoxy)-2-methyl-1-oxopropan-2-yl)oxy)imino)acetate, ATMO3 – 2-(5-amino-1,2,4-thiadiazol-3-yl)-2-(ethoxyimino)acetate
The thioester assay was developed for inhibitors that follow a two-step (reversible bonding, KI, covalent bond formation, kinact) covalent binding mechanism with turnover corresponding to the mechanism of β-lactam inhibition (3). To confirm the results of the first inhibition assay by measuring the hydrolysis of the thioester substrate analog S2d (which could be problematic for other mechanism models because detection is indirect by measuring TNB2–), we decided to additionally perform a fluorescence anisotropy assay (FA) as a secondary biochemical evaluation model (51). Whilst we were only able to determine the residual activity ( RA) of our monocyclic β-lactam compounds in the FA assay, the results generally showed the same trend as observed previously in the thioester assay.
The selectivity and/or reactivity of the synthesized monocyclic β-lactams were then evaluated on other in-house available enzymes ( Supplementary materials, Table SII) that employ catalytic serine, i.e., human butyryl-/acetyl-cholinesterase (hBChE and hAChE, resp.) or cysteine residue, i.e., SARS-CoV-2 main protease, i.e., 3CLpro (42). Suprisingly, ATMO-functionalised monocyclic β-lactams did not inhibit human serine enzymes, suggesting they are likely selective for their bacterial targets. Only some compounds ( e.g., 2, 3, 4, and 8), all possessing a phthalimido moiety at the C3 position, indicated some minimal inhibitory activity in these assays (note that C3 phthalimido intermediates do not inhibit PBP). Next, we wanted to confirm that the detected PBP1b inhibitors were not false positives. Based on our experiences with the detection of false positives due to reactivity with the thiol substrate (Ellman reagent), we tested the prepared compounds in the thiol reactivity assay and successfully confirmed that they are not non-specifically thiol-reactive (46). Furthermore, we confirmed the stability of final compounds in the phosphate buffer at different pH or assay conditions (52, 53). For this purpose, we determined the stability in buffers at different pH values and found that the C3 phthalimido monobactams 1– 8 were unstable even at neutral pH, while on the other hand, the fully functionalized monocyclic β-lactams appeared stable in the phosphate buffer (see Supplementary materials, Table SIII).
Antibacterial activity
We evaluated the antibacterial activity of the monocyclic β-lactams on selected Gram-negative Acinetobacter baumannii 8C6 GES-14 (strain obtained from a European reference laboratory, EURL-AMR, DTU, Copenhagen, Denmark) as a reference strain for process control, Klebsiella pneumoniae RDK 070A (ATCC 51503), Pseudomonas aeruginosa RDK 184 (DSM 939; ATCC 15442)) and Escherichia coli (ATCC 25922), and Gram-positive Staphylococcus aureus (ATCC 29213) and Enterococcus faecalis DRK 057 (ATCC 29212) bacteria. In addition to the potency of the inhibitors against bacterial enzymes, the efficacy in the whole cell assays depends primarily on the success of the uptake of the inhibitors by Gram-positive and Gram-negative bacteria. This depends on the physicochemical properties of the compounds and their ability to be substrates for efflux pumps. Therefore, in order to investigate their uptake profiles, we also determined the antibacterial activity of the compounds using two mutant strains of E. coli N43 (CGSC 5583) and E. coli D22 (CGSC no. 5163), the first lacking an AcrAB efflux pump in its outer cell membrane and the second having a mutation in the lpxC gene that increases membrane permeability. The reference drug was aztreonam.
The C3-phtalimido-protected intermediates 1–8 were inactive, as expected. However, while C3-amido monocyclic β-lactams showed no significant antibacterial activity against Gram-positive wild-type bacteria, moderate antibacterial activity was observed against some Gram-negative wild-type bacteria ( i.e., K. pneumoniae and E. coli, MICs ~ 32–64 μg mL–1, see Table II). Moreover, when the same set of compounds was evaluated against the two mutant strains, interesting results were obtained. While C3-phthalimido analogs 1–8 and the methicillin analog 38 remained inactive, the N1-sulfonate-activated analogs ( e.g., 29, 30, and 33– 36) exhibited moderate antibacterial activity particularly against the E. coli N43 mutant strain with a deleted efflux pump, with MICs ranging from 1–32 μg mL–1 (indicating that these compounds could be AcrA membrane efflux pump substrates), and to a lesser extent in the strain with a mutation in the lpxC gene of E. coli D22 ( MICs 4–32 μg mL–1), compared to wild-type E. coli which was essentially not inhibited. Overall, in the case of the mutant strains, monocyclic β-lactam analog 36 was particularly active ( MICs 1–4 μg mL–1), whereas some other compounds ( 29, 33, 34, 45) also exhibited notable antibacterial activity.
Table II. Antibacterial activities of novel cis -C3/C4 functionalized N -sulfonated β-lactams isolated as TBA+ salts on selected wild-type and mutant Gram-positive and Gram-negative bacterial strains
a Strain with an acrA knockout (cell membrane efflux pump).
b Strain with a mutation in the lpxC gene that increases membrane permeability.
CONCLUSIONS
In this study, we successfully prepared a series of novel C3/C4 substituted N-sulfonated monocyclic β-lactams and evaluated them in vitro. The incorporation of C3 aminothiazole side chains improved the activity of this structural class, as expected based on our previous studies. While none of the compounds were active against a representative Gram-positive strain ( S. aureus), methoxime and the aminooxy-2-methylpropanoic acid derivatives 30 and 35 showed moderate activity against some Gram-negative bacteria ( K. pneumoniae and E. coli strains). The lack of significant in vitro activity of newly developed compounds can likely be attributed to sterically bulkier and highly lipophilic substituents at the C4 position, if compared to aztreonam, which bacteria can expel using efflux pump activity (as proven in the case of mutant strains where our novel compounds exhibited good MIC values). Overall, the results provide a clear scope for further medicinal chemistry optimization of the C4-substituted monocyclic β-lactam class towards analogs which will not be hampered by the activity of efflux pumps, will be sufficiently lipophilic to cross the bacterial outer membrane and will exert potent bactericidal effect on Gram-negative pathogens. Lastly, since β-lactamase-mediated hydrolysis is an important resistance mechanism for this structural class of antibiotics, we will also aim to incorporate β-lactamase testing into our future research efforts. In the follow-up of this work, we will therefore try not only to further optimize the antibacterial activity and cellular permeability of the best-performing compounds but will also simultaneously assess their susceptibility towards clinically relevant β-lactamases to enable an even more informative drug discovery and optimization process.
Abbreviations, acronyms, symbols. – ATMO – aminothiazolemethoxime, CAN – cerium ammonium nitrate, Bn – benzyl, Boc – tert-butyloxycarbonyl, DMB – 2,4-dimethoxybenzyl, Fmoc – fluorenylmethoxycarbonyl, HATU – 1-[bis(dimethylamino)methylene]-1 H-1,2,3-triazolo[4,5- b]pyridinium 3-oxide hexafluorophosphate, hAChE – human acetylcholinesterase, hBChE – human butyrlycholinesterase, LiHDMS – lithium hexadimethylsilazane, MIC – minimum inhibitory concentration, NHS – N-hydroxysuccinimide, PBP – penicillin-binding protein, Ptht – phtalimido, RA – residual activity, TBA – tetrabutylammonium, TBTU – 2-(1 H-benzotriazole-1-yl)-1,1,3,3-tetramethylaminium tetrafluoroborate
Supplementary data. – In the Supplementary materials, synthetic procedures, and spectroscopic data of all compounds described in the manuscript are available. Additionally, selected biochemical and biophysical data are also available.
Acknowledgements. – We thank David Roper laboratory (University of Warwick, UK) for providing E. coli N43 and E. coli D22 mutant strains.
Conflict of interest. – The authors declare no competing financial interest in connection with this manuscript.
Funding. – This research was funded by the Slovenian Research and Innovation Agency (ARIS), Research Core Funding P1-0208, Project N1-0169, Project Z1-4405 and a young researcher grant to K.G.
Authors contributions. – Conceptualization, K.G., N.S.B. and S.G.; synthesis, K.G. and N.S.B.; biochemical and biophysical experiments, K.G., D.K., M.H.R., M.P., K.B. and I.S.; C.C.M. and A.D. provided plasmid and enzyme; data analysis, A.K. and K.G.; writing, original draft preparation, K.G. and A.K.; writing, review and editing, A.K., M.H.R. and S.G.; supervision, S.G. All authors have read and agreed to the published version of the manuscript.