
Introduction
Hereditary diseases such as Epidermolysis bullosa (EB) is caused by mutation of approximately 16 different genes who are involved in the maintenance of the structure and function of dermo-epidermal adhesion in epithelia, and in many cases, it is fatal for patients affected (Wally et al., 2020) (see Table 1 and figure 1).
Skin blister is the main symptoms which characterized Epidermolysis bullosa. Blistering can cause minor trauma and are painful. The prevalence of EB in the United States is 8.2 per million live births (Fine et al., 2004).
The term epidermolysis bullosa (EB) was introduced in 1886 and refers to a group of rare genetic disease which are characterized by varying degrees of skin fragility caused by mutations in the protein level at the various skin structure. There are four main types: Epidermolysis bullosa simplex (EBS), Dystrophic epidermolysis bullosa (DEB), Junctional epidermolysis bullosa (JEB), and Kindler syndrome (KS) (Fine and Hintner 2021)
Intong and Murrell report on recent changes and agree for new definitions during the meeting which is take placed in Vienna, Austria, in Vienna, Austria, in May 2007 and was published in 2012 (Intong and Murrell, 2012).
Table 1. Several types and subtypes of Epidermolysis bullosa.
Based on symptoms the Epidermolysis bullosa (EB) genetically and clinically is characterized by blister formation and erosions of the skin and mucous membranes after minor trauma (Laimer et al., 2009). Mayr et al., (2013) suggest the inheritance of the affected genes can occur in a dominant or recessive way depending on the subform of the disease. The EBs is caused by gene mutations which encode proteins placed in basal membrane zone of the skin. Loss function (absence) of proteins placed in this zone is shown to participate in lacking of akin stability and microarchitecture of the connection between dermis and epidermis leading to a loss of coherence. The connection between dermis and epidermis (called basal membrane) is addicted by keratinocytes and dermal fibroblasts that acts as mechanical support for the connection of both skin layers. The basal membrane also regulates the metabolic exchange between the two skin compartments.
Mayr et al., (2013) report for mutations in the genes, encoding for the keratins 5 and 14 and plectin, lead to epidermolysis bullosa simplex (EBS) characterized by the cytolysis within basal keratinocytes. Loss function of laminin – 332, collagen type XVII or integrin- β4 is shown to cause Junctional epidermolysis bullosa (JEB) which is subtype of EBs. This subtype is the most serve of EBs characterized by separation of skin within lamina lucida. Mutations in type VII collagen (encoded by COL7A1) lead to the dystrophic form of epidermolysis bullosa. The clinical manifestation depended on the mutation type (missense mutation, nonsense mutation, splice site mutation, deletion and insertion).
The aim of this paper is to present the different subtype of EBs and using of different methods for cure of them used and suggested by different research groups.
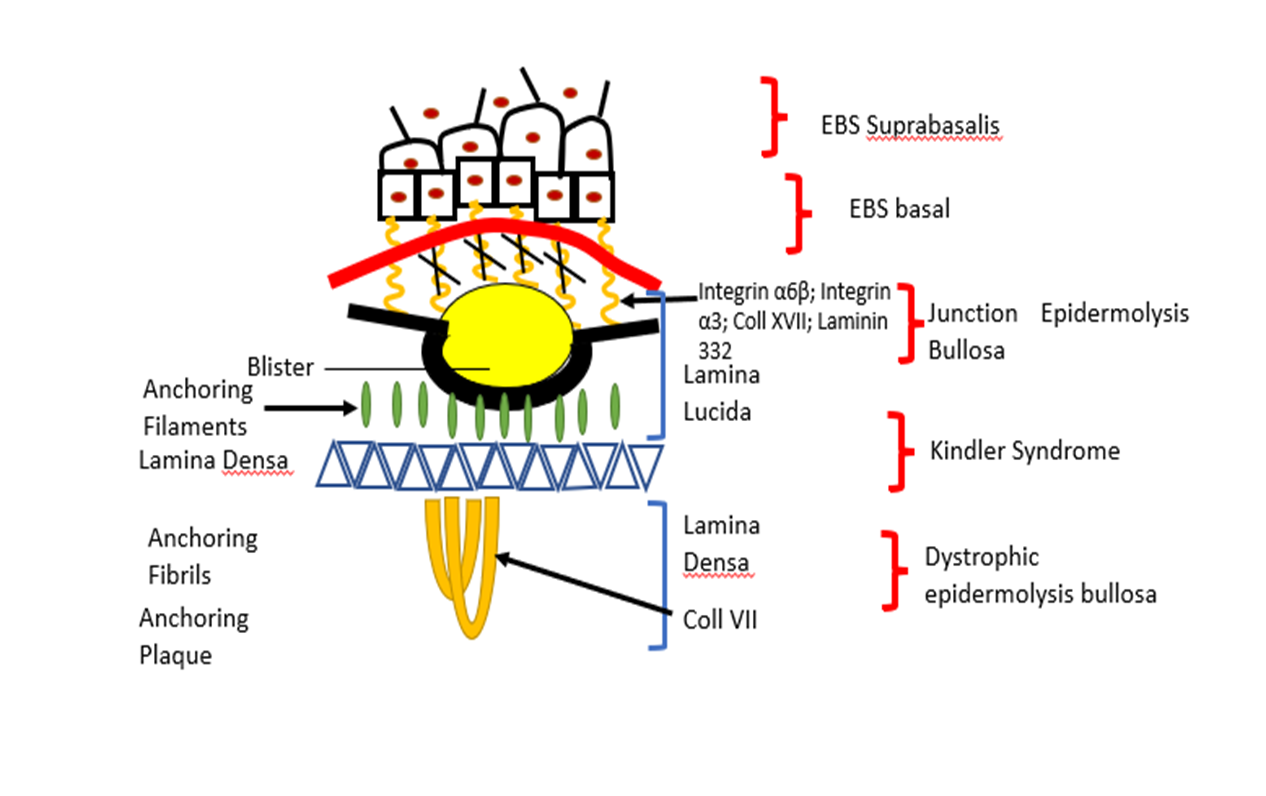
Figure 1. Here are presented different subtypes of EBs and gene mutation which are found.
The therapy suggested for cure of EBs
There is suggestion different type of therapies for EBs cure. For example, gene therapy: correction of JEB by transplantation of epidermis stem cells by Murauer et al., (2015). Using of small molecules such Topical Diacerin in cure of EBs Dowling Meara by Wally et al., (2013). Gene expression studies to identified candidate repair proteins in wound healing suggested by Breitenbach et al., (2015). Bauer et al., (2013) suggest using of specialized ribosome for cure of EBs.
Ribosomal protein has extra function
The ribosome is ribonucleoprotein complex organelle which is responsible for protein synthesis, and its synthesis is highly coordinated; this is shown in involvement of many macromolecules’ components (Temaj et al., 2022; Temaj et al., 2022; Temaj et al., 2022).
Narla and Ebert suggested that several ribosomal proteins have extraribosomal functions, including replication and repair of DNA repair, so mutations in ribosomal proteins may have effects that are independent of the protein translation machinery (Narla and Ebert, 2010).
Which criteria are needed for ribosomal protein to consider that have extra-ribosomal capacity? Warner and McIntosh suggest three criteria: 1) the ribosomal protein in question interacts specifically with some nonribosomal components of the cell, presumably RNA or protein; 2) demonstrating that such interaction in living cell have physiological effects; and 3) evidence that the latter is occurring away from the ribosome (Warner and McIntosh, 2009).
Danilova and Gazda (2015), report that DBA, often is diagnosed during the first year of life; the clinical feature is anemia, low reticulocyte count, macrocytic erythrocyte, increased expression of fetal hemoglobin and elevated activity of adenosine deaminase. Mutation of RPL5/uL18 in DBA caused cleft lips and palats, but this malformation is not observed in mutations of RPS19/eS19 (Gazda et al., 2008).
Mutations of RPL5/uL18 and RPL10/uL16 in DBA have been reported in T-cell acute leukemia (De Keersmaecker et al., 2013). Mutations of 40S subunit ribosomal proteins cause congenital aplesnia (Bolze et al., 2013). In Drosophila melanogaster haploinsuficiency 40S ribosomal proteins is characterized by delay of development, small size, small bristles, and small rough eyes (Marygold et al., 2005). In zebra fishes’ mutations in RPs are associated with delay of development, small size, small head and eyes, brain, apoptosis, reduced pigmentations, pericardial edema and hematopoietic defects (Danilova and Gazda 2015; Amsterdam et al., 2004; Zhang et al., 2014). Mutations of RPS19/eS19 and RPS20/uS10 in mice are associated with dark skin and reduced body (McGowan et al., 2008). RPL24/eL24 mutations in mice lead to the Belly Spot and Tail (Bst) phenotype which is characterized by small size, eye defects, a white ventral spot, white hind feet and various skeletal abnormalities (Oliver et al., 2004). RPL7/uL30 mutation also is manifested with skeletal abnormalities, and with ventral white spot and eye defects (Watkins-Chow et al., 2013).
JEBs (Junction Epidermolysis Bullosa)
The PTC (premature termination codon) in LAMB3 is shown to play pivotal role in JEBs as subtype of EBs. The ribosome as a complex organelle is responsible for translation of LAMB3PTC mRNA aborts protein synthesis at the PTC signal, with production of a truncated, non-functional protein. New drug development in the future must play pivotal in binding with ribosomal protein L35 (rpL35/uL29), and to modified them which will customize increase in production of full-length Lamb3 protein from a LAMB3PTC mRNA. The same authors suggest that Atazanavir and artesunate are the main drug candidate which can bind to ribosomal protein rpL35 and now may tested for their potential to trigger a rpL35 ribosomal switch to increase production of full-length Lamb3 protein from a LAMB3PTC mRNA for targeted systemic therapy in treating JEB (Rathner et al., 2021).
Conclusions
In conclusion, we can say, that in EBs suggested from clinical dermatologist, are used different strategies for cure of epidermolysis bullosa. For example, using of stem cells are shown to be very benefits in some subtypes. Employment of small molecules such as drug diacerin for treatment of EBs Dowling Meara. Gene technology for correction of mutated gene COL7A1 by trans-splicing in dystrophic epidermolysis bullosa (DEBs). Last time is suggested modification of ribosomal protein for translation repair of PTC (premature termination codon) in mutated JEBs.