
INTRODUCTION
Remifentanil is a synthetic opioid-class analgesic with an ultra-short duration of action. It is given intravenously and is very quickly cleaved by plasma- and tissue-esterases to inactive remifentanil acid, resulting in an extremely short remifentanil biological half-life of only 3–10 min (1). It is being used as an analgesic for pain management during anesthesia, and in intensive care units because of its rapid initiation and subsequent termination of the action. It is also being increasingly used “off-label” as pain management during labour. The recent draft guidelines by the National Institute for Health and Care Excellence (NICE) recommend remifentanil for parturients who do not want epidural anaesthesia (2). For this purpose, a patient-controlled analgesia system (PCA) is often used, where the parturient can administer bolus doses from 0.15 up to 0.9 µg kg–1 of remifentanil before each contraction with or without concomitant basal infusion (of up to 0.05 µg kg–1 min–1) (3–5). However, the recent protocols usually include bolus doses of 20 to 40 µg, depending on parturient weight, with one to two-minute lockout periods (6, 7).
Adverse drug reactions may include dose-dependent and plasma concentration-dependent hypotension, bradycardia, muscle rigidity, and respiratory depression. Blood concentrations of 1–3 ng mL–1 are considered analgesic with minimal (but nevertheless possibly present) effects on ventilation. Spontaneous ventilation occurs at 4–5 ng mL–1 (1). A study on remifentanil PCA administration during labour reported maternal remifentanil plasma levels of approximately 5 ng mL–1 (8). According to the manufacturer, when the concentration exceeds 5 ng mL–1, transient respiratory depression, apnea, and muscle rigidity can occur (1). Furthermore, remifentanil rapidly crosses the placenta, and its pharmacokinetic and pharmacodynamic profile may be different from that of an adult (9).
Nevertheless, the theoretical and reported growing practical evidence speaks in favour of safe remifentanil use during labour if the established standard procedure is followed, as was recently reported by the authors from RemiPCA SAFE Network (5). Although many obstetric clinics already routinely offer an option of remifentanil as the most effective alternative technique to epidural analgesia (10), a clear consensus about its safe applicability for this indication has not been reached (11, 12), and additional research is necessary to determine the safety of remifentanil treatment during labour (13).
Even though the parturient heart and ventilatory function or saturation must be constantly monitored by an anaesthesiologist, some cases of respiratory arrest in mothers have been associated with remifentanil use during labour (14–18). Moreover, the short- and long-term effects on infants caused by remifentanil use during labour have not been fully characterised either (19, 20).
Therefore, more trials are needed to better elucidate remifentanil pharmacokinetic and pharmacodynamic profiles in both the mother and the child to provide the optimal dosing regimen in terms of reaching the best compromise between efficacy and safety.
To support such trials, a suitable analytical method for remifentanil blood levels is required, where the analyte stability is not compromised after blood withdrawal due to hydrolysis of the ester bond by plasma esterases in obtained samples (Fig. 1). An additional requirement is that the method would require only small volumes of blood samples, suitable for neonatal monitoring. Ideally, the sampling could be obtained as dried blood spots, since this is a long-established routine technique for neonatal screening. Furthermore, the dried blood spots can be obtained from umbilical cord blood, minimizing the burden for the newborn, and at the same time, getting also the most relevant sample for determination of true remifentanil concentration in the newborn at the time of cord clamping. Recently, a method for the determination of 132 psychoactive compounds in dried blood spots has been reported, which included also remifentanil (21), but with a limit of quantitation of 5 ng mL–1, it lacks the sensitivity needed to assess the levels possibly leading to the adverse drug effects in newborns and their mothers, which can start to occur already at 1–3 ng mL–1.
Accordingly, the aim of our study was to develop a rapid and sensitive method for the determination of remifentanil in dried umbilical cord blood spots to support the efficacy and safety trials of remifentanil during labour, with a limit of quantitation below the threshold of 1 ng mL–1, where the adverse effects may start to appear.
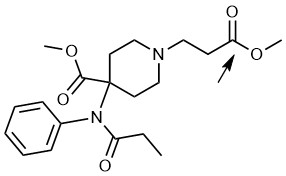
Fig 1. The chemical structure of remifentanil, with the arrow pointing to the ester bond subjected to enzymatic cleavage during its rapid elimination from blood.
EXPERIMENTAL
Remifentanil and isotopically labelled internal standard remifentanil-13C6 (IS) have been purchased from Toronto Research, Canada.
Blank dried blood spot cards Whatman 903 have been obtained from GE Healthcare Bio-sciences Corp. Piscataway, NJ, ZDA.
Blank pooled human blood and plasma for method development were obtained from the University Clinical Centre, Ljubljana, Slovenia.
The study has been approved by the Slovenian National Medical Ethics Committee (Grant Nr.: 96/10/09).
The study was conducted at the Gynaecology and Obstetric Hospital Kranj. A written informed consent was obtained from all participants. Inclusion criteria were: age above 18 years, expressed request for pain relief other than epidural analgesia, gestation period of at least 35 weeks, body weight 50–100 kg, spontaneous or induced delivery, singleton term pregnancy, healthy status of parturient and fetus with normal cardiac function and cephalic presentation. Exclusion criteria were: allergy to opioid analgesics, age below 18 years, diabetes mellitus, pre-eclampsia, heart, liver, or kidney disease, twins, intrauterine growth retardation with estimated foetus weight below 2500 g, pathological cardiotocography (CTG), and fetal distress from whatever reason.
Two parturients (and their two neonates after delivery) were enrolled in the study. The maternal arterial blood in Case 1 was sampled just after the birth by direct arterial puncture. In Case 2 maternal blood sampling was omitted due to obstetric intervention after birth. All umbilical arterial and venous blood were sampled immediately after cord clamping. All samples were collected in standardized syringes for arterial blood gas analysis with anticoagulant (2.2 mL).
The remifentanil administration was in case 1, bolus doses of 0.41 µg kg–1 with a basal infusion of 0.026 µg kg–1 min–1. In case 2, the bolus doses were 0.31 µg kg–1 with basal infusion of 0.019 µg kg–1 min–1. The bolus doses were administered over 30 s intervals with 2 min lockout periods. The PCA was stopped at the beginning of the expulsion phase and vital functions were constantly monitored. The time from PCA termination until the delivery was noted as well as the time between the delivery and cord clamping with blood sampling.
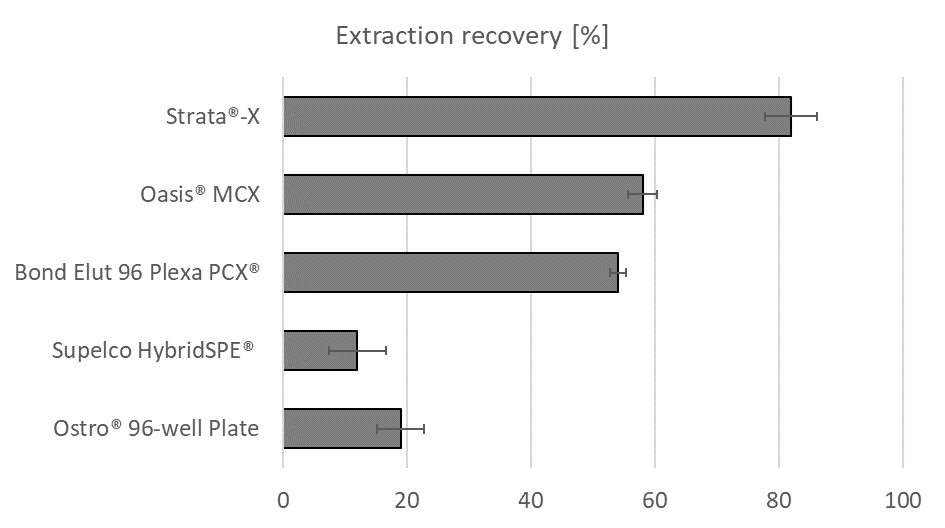
Fig. 2. Remifentanil recoveries (with RSD) obtained during the testing of different sorbents for the optimization of the sample preparation procedure.
Liquid chromatography-tandem mass spectrometry (LC-MS/MS)
The reconstituted samples were subjected to analysis by UHPLC Agilent 1290 coupled to an Agilent 6460 triple quadrupole mass spectrometer. 3 µL were injected onto a 100 × 3 mm Kinetex C18 column (2.6 µm particles) at 50 °C and eluted with mobile phase A (0.1 % formic acid in water), and B (acetonitrile) at 0.65 mL–1 min using the following linear gradient (time [min], %B): (0,10); (0.25,10); (1.25,20); (2.40,30); (2.8,50); (3.4,90); (4.0,10). The total run time was 5.4 min. For MS detection, an ESI JetStream interface in positive mode was used. The following mass transitions (MRMs) were monitored (parent → daughter m/z; collision energy): remifentanil: 377.2→317.2; 8 eV), IS: 383.4→113.0; 25 eV) (Fig. 3).
Method validation
The method was checked according to the recommendations for validation of DBS methods (27) and included the following validation parameters: accuracy, precision, selectivity, extraction recovery, absolute and relative matrix effect, linear range, the lower limit of quantification, and stability under the predicted sample storage conditions.
RESULTS AND DISCUSSION
The method has been successfully developed and validated according to the purposes of our study.
Selectivity
The selectivity was determined by visual inspection of 6 processed blank matrix samples, where no interfering peaks were evident, apart from clearly observable, but not considered as problematic, chromatographic system-related carry-over of approximately 0.4 %. Nevertheless, even such a low carryover can represent up to 30 % of the response of the LLOQ sample (Fig. 3) when a blank extract is injected after a QC-high sample. However, this carry-over was not considered problematic since it is easily solved by a double sample injection.
Sensitivity and linear range
The limit of quantitation was established at 0.3 ng mL–1, where good accuracy of 92 %, as well as acceptable precision of 18 % RSD, and a signal-to-noise ratio above 20:1 was achieved. The method response showed good linearity on a wide concentration range from 0.3 to 40 ng mL–1 with a high determination coefficient ( R2 > 0.999).
Accuracy, precision, recovery, and matrix effect
Assay accuracy, precision, recovery, and matrix effect data are presented in Table I, and the calibration plot in Fig. 4. Absolute matrix effect and recovery were determined by comparison of instrumental responses obtained from neat solutions (A), post-spiked (B), and pre-spiked (C) samples at concentration levels of QC-low, and QC-high samples. The absolute matrix effects, calculated as (B/A-1)*100 %, and recovery (C/B*100 %), were repeatable and comparable between high and low QC samples (Table I). The relative matrix effect was determined to be insignificant as the variability of the regression line slopes obtained in six different lots of blood, was 3.5 % RSD; an acceptable value is under 3–4 % RSD (22), therefore the method can be considered as free from significant relative matrix effects, which was expected due to the use of the isotope-labelled internal standard.
Table I. Accuracy, precision, recovery, and absolute matrix effect determined on QC samples
Stability
The stability testing of DBS samples stored at various conditions has shown less than 10 % deviation from their initial values which indicates that the analyte is stable at all three tested temperatures (–20, 24, and 40 °C) for at least a period of 14 days. The reported remifentanil half-life in blood ex vivo is 99 ± 6 min (23, 24), therefore the stability of blood samples before being applied onto the pre-treated DBS cards should not be considered problematic if the sample is applied to a pre-treated blank DBS card with citric acid immediately after blood withdrawal or at least in a reasonably short time thereafter . Indeed, by testing consecutively prepared DBS samples within 4 minutes in 30 s intervals for each QC level we have not detected any observable analyte peak response deterioration. The stock solution and autosampler stability studies indicated that remifentanil and IS solutions were stable for at least 48 hours at 4 °C.
DBS-specific validation parameters
Some of the recommended validation parameters according to the recent DBS-related guidelines were not determined because they were not applicable to the present study (25, 26). For example, the spot homogeneity, and effects of the spot volume and haematocrit were not tested because the blood samples were applied using a repeater pipette (with a fixed volume of 20 µL) and after drying, the whole spot was excised, thereby eliminating all issues regarding the spot concentration or inhomogeneity (27). Furthermore, the effect of haematocrit on analyte recovery could not be performed due to the rapid analyte breakdown in blood incubation conditions usually employed for such a test (gentle shaking at 37 °C, 30–45 min). Even though the neonates are known to have higher and variable haematocrit (28), minimal effects of differences in haematocrit on remifentanil recovery are expected since the red blood cell-to-plasma ratio for remifentanil is 0.89, which is close to unity, therefore a change in haematocrit would not lead to any significant differences in remifentanil partitioning (29, 30). Furthermore, since our results show high recovery from the whole blood spots (Table I) and comparable recovery from dried plasma spots (data not shown), also the extraction recovery should remain unaffected with varying haematocrit.
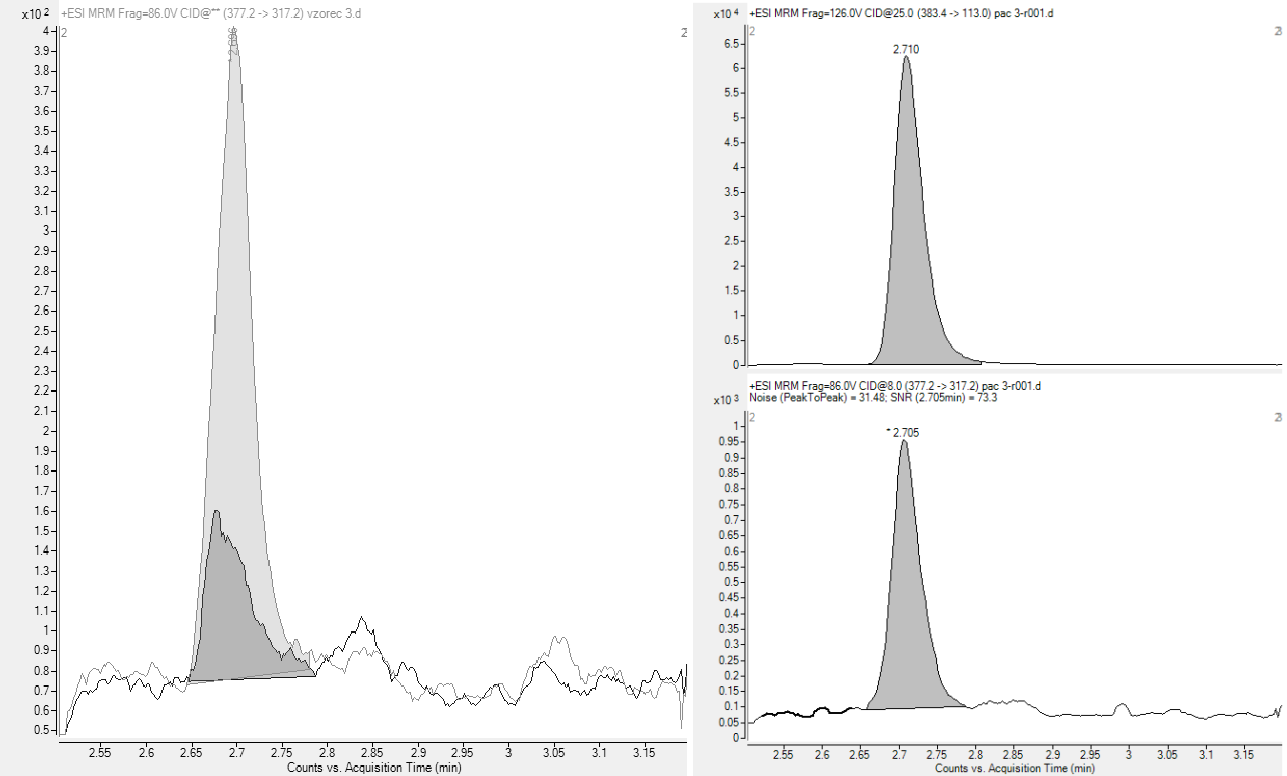
Fig. 3. Left-hand side: A representative SRM chromatogram of an LLOQ sample (light grey trace) compared to an extracted blank sample (dark grey trace). Right-hand side: A representative parturient sample with MRM chromatograms for remifentanil-13C6 (top) and remifentanil (bottom).
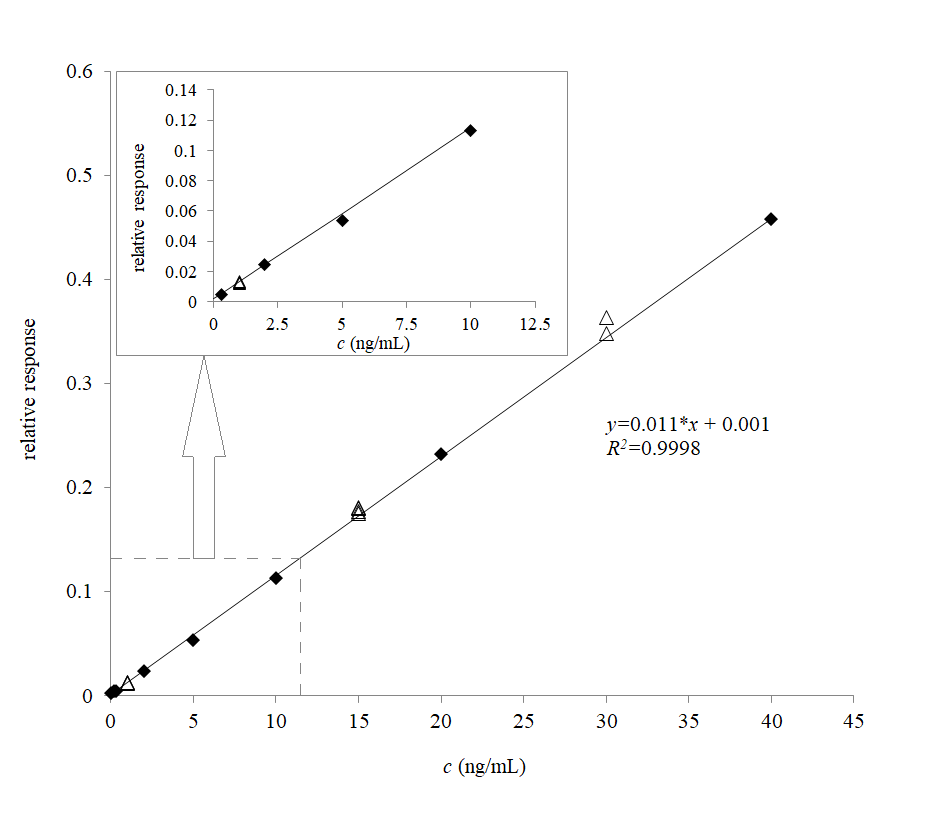
Fig. 4. The calibration line for remifentanil in human dried blood spots; the relative response (y-axis) is the ratio between the peak area of remifentanil and that of13C6 remifentanil (IS). Closed diamonds represent the calibrators, while the open triangles represent the QCs; weight: 1/c. The enlarged section outlined by the dashed line from 0 to 12 ng mL –1demonstrates linear response down to the lower limit of quantitation.
Table II. The determined and estimated remifentanil concentrations in maternal and umbilical cord blood samples
a Estimated value, due to the concentration being below the limit of quantitation (0.3 ng mL–1).
When comparing our results to a similar study by Shen et al. (8), where the mean maternal arterial remifentanil concentration in the PCA group was 4.95 ng mL–1, and the mean umbilical venous concentration was 2.98 ng mL–1, a significant difference to our findings can be observed. At first, the disagreement between the results seems difficult to explain, since the bolus doses in PCA groups in both studies were similar: 0.31–0.4 µg kg–1 administered over 30 s with 2 min lockout periods, which can be regarded as comparable, especially when considering also the basal infusion in our study 0.019–0.026 µg kg–1 min–1 (8). However, the main difference between our results (Table II) and the above-cited concentrations can be attributed to dissimilarity in the duration of the PCA remifentanil administration relative to the delivery and blood sampling time point. In our study, the PCA was stopped at the beginning of the expulsion phase, whereas in the study by Shen et al. (8), the PCA had been enabled throughout the expulsion phase until the delivery, and the time from the last bolus to sampling was only 74 s in average, which coincides with the estimated remifentanil peak blood concentration (8). Therefore, in our study, the observed significantly lower maternal and neonatal remifentanil concentrations after delivery can be attributed predominantly to the longer period without PCA before the expulsion phase, delivery, and blood sampling. The cumulative amount of time after the last bolus until our sampling would sum up to 5–12 minutes (Table II), enough to account for the significant remifentanil concentration drop with the context-sensitive half-life below 3.5 min. The lower concentrations at the time of delivery would also mean a safer and more conservative pain management strategy due to the lower probability of adverse events, especially in the neonate, but in the mothers as well.
Finally, the presented method, albeit applied only to a very limited number of samples in our small pilot study, has been proven sensitive enough for the safety assessment of newer remifentanil administration protocols employing only 0.4 µg kg–1 bolus doses. The found concentration levels were all below the threshold of expected adverse drug effects of 1–3 ng mL–1, even when extrapolated to the time of delivery (Table II). Our results go hand-in-hand with the recent findings of a large meta-analysis which compared the remifentanil PCA with epidural analgesia during labour and found no differences in Apgar scores < 7 at 5 minutes in both groups, although the incidence of respiratory depression was nevertheless significantly higher in the PCA group (33). It is important to note that our analytical method can measure remifentanil in a wide concentration range, from the sub-therapeutic 0.3 ng mL–1 to toxic 40 ng mL–1. The large working range may be useful also for other remifentanil applications, not only for labour analgesia. Significantly higher concentrations compared to our pilot study can be expected also in other remifentanil PCA protocols as demonstrated in the study by Shen et al. since in some practices, and possible future efficacy/safety studies, the administration of remifentanil is used or expected to last through the whole delivery, particularly throughout the expulsion phase, where effective pain control is also most needed and desired, resulting in much higher blood concentrations of remifentanil at the time of delivery. This applies to neonatal and maternal samples whose concentrations are expected to be approximately two-fold higher than that of the neonates, and hence, the mothers might be at higher risk of serious adverse events (1, 18, 33), underlining the need for appropriate dose-finding studies combined with individualization based on clinical assessment. Other venous and capillary blood sampling methods could also be used in conjunction with our analytical methods, such as volumetric absorptive microsampling (VAMS) (34), finger-prick (35), and heel-prick sampling (36).
Acknowledgments. ‒ The authors are thankful to Mr. Erik Tičar, MPharm., for his help with the sample preparation.
Funding. ‒The study has been supported by the Slovenian Research Agency (ARIS) (P1-0189).
Conflicts of interest. ‒ The authors declare no conflict of interest.
Authors contributions. – Conceptualization, J.T., A.R., and A.M.; methodology, J.T., A.R., and A.M.; investigation, A.R.; analysis, J.T.; writing, original draft preparation, J.T., A.R., and A.M.; writing, review and editing, J.T., A.R., and A.M. All authors have read and agreed to the published version of the manuscript.