
Introduction
Benzimidazoles are veterinary drugs widely used for the prevention and treatment of parasitic infections in agriculture and aquaculture, including food producing animals (Danaher, 2007). The extensive use of benzimidazoles in lactating animals could result in the presence of their residues in milk and dairy products, which is harmful to consumers owing to their teratogenic and embryotoxic properties (Chen et al., 2010.). To ensure human food safety, the European Union set a maximum residue limits (MRLs) in animal tissues for most benzimidazoles, though only albendazole, fenbendazole, triclabendazole and thiabendazole have MRL values in milk (EC, 2010;EC, 2012;EC, 2014). The marker residue of almost all the benzimidazoles is defined as the sum of a parent drug and/or its metabolites.
The extraction and purification of benzimidazole residues from milk poses a difficult challenge. At present, the majority of methods used for detection of benzimidazole residues from milk do not generally include the complete range of marker residues for each substance. For effective control, benzimidazole residues in foods, including milk, methods capable of detecting the complete range of marker residues should be applied. In recent years, liquid chromatography coupled with tandem mass spectrometry (LC-MS/MS) has found more widespread application in benzimidazole residue analysis, offering more sensitive detection and increased confidence in reporting results (DeRuyck et al., 2002;Jedziniak et al., 2009;Xia et al., 2010;Chen et al., 2011). However, mass instruments are still quite expensive and not readily available to chemists in most laboratories. HPLC is the most widely applied method to separate residues of benzimidazoles drugs. Although fluorescence is more sensitive and selective, the most widely applied detector is ultraviolet (UV) (Takeba et al., 2000;DeRuyck et al., 2000).
The development of methods that cover the complete number of marker residues or the most persistent or predominant metabolites is difficult. As a result, a number of important benzimidazole residues, such as triclabendazole (TCB) and its metabolites, are not included into multi-residue methods. To date, there is only one published multi-residue method that encompasses 11 benzimidazoles and 10 metabolites in milk using the HPLC-UV technique (Chen et al., 2010). Disadvantages of that method are the long analysis time for HPLC-UV, which takes 80 min, and the fact that it does not include one of the triclabendazole metabolites - ketotriclabendazole. The aim of this study was to develop a HPLC multi-residue method with DAD detection for the determination of benzimidazole markers and their metabolites in milk. The developed method was applied to the analysis of real milk samples taken from dairy farms in Croatia.
Experimental
Drugs and reagents
Analytical standards of albendazole sulphoxide (ABZ-SO), albendazole sulphone (ABZ-SO2), albendazole-2-amino-sulphone (ABZ-NH2-SO2), hydroxy mebendazole (MBZ-OH), amino mebendazole (MBZ-NH2), triclabendazole sulphoxide (TCB-SO), triclabendazole sulphone (TCB-SO2), and ketotriclabendazole (KTCB) were purchased from Witega (Berlin, Germany). Mebendazole (MBZ), fenbendazole (FEBZ), fenbendazole sulphoxide (oxfendazole, OFZ), fenbendazole sulphone (FEBZ-SO2), thiabendazole (TBZ), 5-hydroxy thiabendazole (5-OH-TBZ), flubendazole (FLU), 2-aminoflubendazole (FLU-HMET), oxibendazole (OXI) and nocodazole (NOC) as the internal standard were purchased from Sigma Aldrich (St. Louis, MO, USA).
Chemicals of analytical grade including glacial acetic acid, ammonium acetate, ammonium hydroxide, hydrochloric acid, anhydrous sodium sulphate, dimethylsulfoxide (DMSO), n-hexane and ethanol were supplied from Kemika (Zagreb, Croatia). Acetonitrile and methanol were HPLC-grade and also obtained from Kemika (Zagreb, Croatia). Water was purified with a Millipore DirectQ5 UV system (Merck Millipore, USA).
The ammonium hydroxide-acetonitrile solution used for the eluation of analytes was prepared on the day of use by mixing 160 mL acetonitrile with 22 mL conc. ammonium hydroxide and making up to 200 mL with acetonitrile.
The cartridge used for solid phase extraction (SPE) was PLEXA PCX cartridge (500 mg, 6 mL, Agilent, Milford, HA, USA).
Prior to HPLC injection, samples were filtered through a 0.45 µm PTFE syringe filter from FilterBio (Nantong City, Jiangsu P.R China).
Preparation of stock and working solutions
Individual stock standard solutions (1 mg/mL) of all analytes were made by dissolving each standard in DMSO and stored at -20 °C for 5 years.
For linearity testing, a mixed standard solution containing 20 or 50 µg mL-1 of all compounds was prepared by combining 200 or 500 µL of each standard stock solution and diluting to 10 mL with ethanol. For other validation parameters study, a mixed standard solution containing 2.5 or 5 µg mL-1 was prepared. The solutions were stored at -20 °C for 1 year. During these periods we have not observed any signs of decomposition of analytes. Standard working solutions, at various concentrations, were prepared daily by the appropriate dilution of the mixed standard solution with mobile phase A.
Sample collection
Cow milk samples were collected by authorized veterinarians at dairy farms in different regions of continental Croatia. In total, 50 milk samples were sampled in different counties: 7 in Zagreb County, 8 in Krapina Zagorje County, 8 in Karlovac County, 8 in Koprivnica Krizevci County, 6 in Brod Posavina County, 13 in Baranya County. Milk samples were collected in a polyethylene bottle of 50 ml volume. Samples were stored at -20 °C until the time of analysis. Analysed milk samples that were found to contain no detectable residues of the analytes were used as the negative controls (blank).
Sample preparation
A 10 g sample of milk was weighed into a 50 mL centrifuge tube and 2-3 g anhydrous sodium sulphate was added. The mixture was vortexed for 1 min and 10 mL acetonitrile was added. This mixture was shaken for 10 min and centrifuged for 15 min at 3600 rpm. The supernatant was decanted into another 50 mL centrifuge tube containing 10 mL water and 10 mL 0.1 M hydrochloric acid. This mixture was washed with 10 mL hexane (shaken for 10 min, centrifuged for 5 min at 3000 rpm), the hexane phase was discarded and the rest was diluted with water to 50 mL.
The PCX cartridge was pre-conditioned with 5 mL methanol and 5 mL water. Entire extracts were loaded onto the SPE. The column was washed twice with 5 mL 0.1 M hydrochloric acid and then with 5 mL methanol. The column was then dried by purging air at 600 mbar for 10 min. The analytes were eluted with 10 mL ammonium hydroxide-acetonitrile solution, evaporated to dryness under nitrogen at 40 °C and redissolved in 40 µL methanol, vortexed for 1 min, with the addition of 160 µL mobile phase A, vortexed for 1 min and the solution was filtered before HPLC analysis.
Instrumentation and chromatographic conditions
Benzimdazole determination were performed using HPLC-DAD system comprising of 1260 degasser, 1260 dual pump system, 1290 autosampler, 1260 column oven and 1200 DAD detector (Agilent, Palo Alto, CA, USA). Gradient elution using column Xbridge C18 4.6 x 150 mm, 3.5 μm equipped with a guard column 3.5 µm, 4.6x20 mm was used for chromatographic separation (Waters Corp., Milford, MA, USA). The column temperature was maintained at 55 °C in gradient mode with buffer solution containing 0.01 M ammonium acetate to pH 5.7 with 1 % acetic acid as mobile phase A and acetonitrile as mobile phase B. The gradient program was: 0 min 82 % A, 20 min 67 % A, 25 min 10 % A, 26 min 82 % A. The flow rate during analysis was 1 mL/min. The injected volume was 50 µL DAD and the analysis was performed monitoring four different wavelengths: 298, 312, 254 and 290 nm with scan the entire spectrum from 190 to 320 nm.
Method validation
The developed method was validated according to procedures described in Commission Decision 2002/657/EC covering specificity, calibration curve linearity, precision, decision limit (CCα) and detection capability (CCβ) (EC, 2002).
In order to determine validation parameters, different amounts (100, 200 or 300 µL) of working standard solution (0.5 respectively 5 µg mL-1) were added to 10 g samples of bovine and ovine milk free from benzimidazoles. The samples were held at room temperature for 15 min prior to extraction ensuring absorption of the standard solution. The results of validation were calculated for individual substances.
Selectivity. To establish the selectivity/specificity of the method, milk samples fortified with the benzimidazoles at MRLs and at 5 µg kg-1 for substances without MRLs and non-fortified samples were analysed to verify the absence of interfering substances around the retention time of analytes
Linearity and quantitative analysis. Standard calibration curves were prepared with the injection of working standard solutions at six concentration levels from 0.125 to 4 µg mL-1 respectively from 0.3125 to 10 µg mL-1 and calculated using linear least squares regression analyses of the peak area to concentration ratios.
Precision and accuracy. Toestimate the precision of the analytical method, blank milk samples were fortified with a standard working solution (0.5 respectively 5 µg mL-1) containing a mix of benzimidazoles at four levels for substances with MRLs (5, 10 and 15 µg kg-1; 25, 50, 100 and 150 µg kg-1) and at three levels for substances without MRLs (5, 10 and 15 µg kg-1) in six replicates for each concentration. The complete extraction and purification procedures were performed for the spiked samples. Within-laboratory reproducibility was determined by repeating the study on three consecutive days with two different analysts and different lots of chemicals. For each level, the average concentration, standard deviation (SD), coefficient of variation (CV, %) and recovery were calculated.
CCα and CCβ. Decision limit (CCα) and detection capability (CCβ) were calculated using within-laboratory reproducibility validation results, according to the equation:
- for substances with MRLs
CCα = MRL + 1.64 × SDWLR
CCβ = CCα + 1.64 × SDWLR
MRL- concentration equal to MRL value; SDWLR - standard deviation of within-laboratory reproducibility,
- for substances without MRLs
CCα = c0 + 2.33 × SDWLR,C0
CCβ = CCα + 1.64 × SDWLR,C0
c0- concentration equal to the lowest spiking level; SDWLR,C0 - standard deviation of within-laboratory reproducibility at c0
LOD and LOQ. Limit of detection (LOD) was calculated on the basis of the lowest concentration according to the equation:
LOD = SDWLR,C0 × t0,02
LOQ = 10 × SDWLR,C0
t0,02 - student t value at n-1 degrees of freedom and probability of 98%; SDWLR,C0 - standard deviation of within-laboratory reproducibility at c0.
Statistical analysis of data
Statistical analysis of the results data of validation parameters was performed using the Microsoft Excel program. Calculated statistical values included the mean, standard deviation (SD) and coefficient of variation (CV %). Results data were analysed with statistical method Anova Single Factor with level of significance p=0.05.
Results and discussion
In this study, we were looking for a simple, fast and reliable method for the determination of benzimidazole drugs. Due to the great difference in chemical properties, it was difficult to develop a method to cover the entire range of benzimidazoles. Acetonitrile and ammonium acetate buffer were selected as the mobile phase according toChen et al. (2010). The maximum sensitivity and satisfactory separation of all analytes was achieved when using 0.01 M ammonium acetate (pH 5.7 with 1 % acetic acid). The gradient was optimized to provide the maximum separation possible in minimum time period. The biggest challenge was to separate all triclabendazole metabolites, since they elute very closely. Column temperature, in addition to the concentration of acetonitrile in the mobile phase, proved to be an important separation factor.
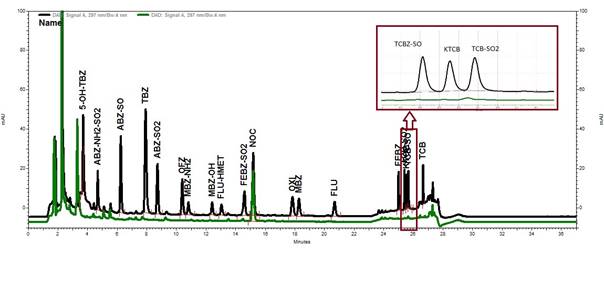
No interfering peaks were observed for the retention time for benzimidazoles, except for the retention times for the triclabendazole metabolites and FEBZ-SO2 (Fig. 1) where an interfering peak of negligible size, relative to MRL concentration, were observed.
The linearity of standard calibration curves was acceptable and within the range from and values of R2 for all analytes were above 0.99.
Mean recoveries (n = 6) of the analytes, determined in six separate assays, were between 60 % and 100 % (Table 1). Lower mean recoveries of approximately 30 % were obtained for KTCB and TCB-SO2, higher mean recoveries of approximately 120 % were obtained for ABZ-SO2 and 135 % FEBZ-SO2. These obtained values were not acceptable according to Commission Decision 2002/657/EC, but since the statistical analysis ANOVA showed no differences among the results, and because of comparability with the results of other studies, obtained results were accepted in that kind with obligatory correction when expression results. The procedure did not prevent the oxidation of benzimidazoles. This is especially notable in the case of high recoveries of FEBZ-SO2. Similar low values for recoveries (21-33 %) and low precision of determination (CV 22-34 %) of FEBZ together with high recoveries of FEBZ-SO and FEBZ-SO2 were found in study ofJedziniak et al. (2009). The authors described sample preparation as extracting analytes from an alkalized matrix to ethyl acetate and then defatting the extract with hexane. To prevent the oxidation of benzimidazoles, two antioxidant agents were used. TCB metabolites were recovered at approximately 30-50 %. Although authors used different methods of sample preparation comparing to our method, obtained similar validation results indicate further need to develop methods of sample preparation with the aim of better results for validation parameters of named analytes. Another possibility is expressing validation results for the sum of parent compounds and their metabolites which showed more reliable accuracy and precision than for individual substances (Jedziniak et al., 2009.).Danaher et al. (2007) indicated that TCBZ metabolites are in a neutral state at a lower pH than other benzimidazoles and that this can have an influence on the repeatability of the extraction of these compounds. In a similar study using liquid–liquid extraction, sample preparation with acetonitrile, with an additional solid-phase sample extraction step procedure, followed by quantification by HPLC-UV method, the recoveries were determined in the range from 78 % to 109 % (Chen et al., 2010). The reported method did not include the metabolite KTCB. To quantify the target analytes in milk, the authors used matrix-matched calibration curves within the range from 10 to 200 µg kg-1. The validated method described in this study covers the range from 2.5 to 200 µg kg-1, which is a much lower concentration range. In another study on the determination of anthelmintic compounds, including benzimidazole compounds, using the QuEChERS procedure for sample preparation and detection by UPLC-MS/MS, a recovery of 70-110 % was obtained (Whelan et al., 2010).
The results of accuracy and precision data are summarized inTable 1. For most analytes, CV% values were less than 26 %. In some instances (TCB metabolites), CV% values were in excess of 25 %. CV theoretical was calculated according to the Horowitz equation CV% = 2 1-0,5logC and compared to the validation results data. In the case of not matching results data were analysed with the statistical method Anova Single Factor (p=0.05), which found no significant differences among the results.Chen et al. (2010) showed intermediate precision expressed as RSD % in the range 4-16 %. Comparing all analyses, higher values were obtained for TCB and its metabolites. The data reported in that study were obtained in the analysis of the corresponding samples at 10, 50 and 100 µg kg-1, which was only partially comparable to the present study since validation results for substances without MRLs were obtained at lower concentrations: 5, 10 and 15 µg kg-1. In another study using the QuEChERS procedure for sample preparation, CV% for repeatability was >23 % for TCB-SO, TCB-SO2 and 5-OH-TBZ at low level validation (2, 3 and 4 µg kg-1) (Whelan et al., 2010).
*Average measured values ± SD
In the present study, low values were obtained for LOD (1-4 µg kg-1) and LOQ (4-18 µg kg-1). Sensitivity of the method was far below the MRL levels.Chen et al. (2010) obtained a similar LOD and LOQ values of 3 and 10 µg kg-1 using HPLC-UV. However, in another study using ultra high performance liquid chromatography and tandem mass spectrometry (UHPLC-MS/MS), LOD values were much lower: 0.1-0.6 µg L-1 (De Ruyck et al., 2002) and <1 µg kg-1 (Whelan et al., 2010).
The CCα and CCβ values are summarised inTable 2. Obviously, the highest percentile deviation on the target concentration was obtained for analytes without an MRL and analytes with a lower value of the MRL (10 µg kg-1). There are very few reported data on CCα and CCβ values. The CCα and CCβ values determined using UHPLC-MS/MS analysis were lower than those reported here, and ranged from 0.14 to 1.9 and 11 to 123 µg kg-1 for unapproved and MRL substances, respectively (Whelan et al., 2010).
Application of the method
The described analytical method was applied to real samples to assess the occurrence of anthelmintic drugs. In the present study, 50 samples of raw milk were collected in Croatia during 2014 and analysed. No detectable residues of the target analytes were found in any of these samples.
Conclusions
This paper reports the development and validation for a HPLC–DAD method for the simultaneous determination of following benzimidazoles and their metabolites: albendazole sulphoxide, albendazole sulphone, albendazole-2-amino-sulphone, hydroxy mebendazol, amino mebendazole, triclabendazole sulphoxide, triclabendazole sulphone, ketotriclabendazole, mebendazole, fenbendazole, fenbendazole sulphoxide, fenbendazole sulphone, thiabendazole, 5-hydroxy thiabendazole, flubendazole, 2-aminoflubendazole and oxibendazole. Benzimidazoles compounds were successfully separated by HPLC. The method has satisfactory validation characteristics according to Commission Decision 2002/657/EC for almost all analytes, except for KTCB, TCB-SO2, FEBZ-SO2 and ABZ-SO2. Statistical analysis of the validation results have not observed any significant differences within results, so with obligatory correction of recovery when expressing results, method is suitable for the routine determination of benzimidazoles residues in milk. Further development of method in step of sample preparation in order to get a satisfactory validation parameters of listed analytes should be taken or validation for the sum of parent compounds and their metabolites should be carried out.