
INTRODUCTION
Doxorubicin (Dox) is one of the most used anthracyclines in oncology treatment for different types of cancer in combination with other antineoplastic drugs (1, 2). Despite its side effects causing complications after chemotherapy is done and the patient is free of cancer, it has been approved globally for several decades now. The main mechanism of Dox side-effect is related to oxidative damage on the cell level which can be prevented by co-treatment with strong antioxidants (3). One of them which has been intensively investigated for the past 15 years is the water-soluble form of fulleren C60, the so-called fullerenol. Fullerenol form, C60(OH)24, as a strong antioxidant, was mostly investigated in acute and chronic Dox-induced toxicity, to protect organs against long-term damage (3). Its protective role was demonstrated in the heart, liver, lung, kidney, and testes, in rats (1, 3, 4–6).
From the analytical point of view, there are many articles covering Dox chromatographic routine tests and control methods as well as pharmacokinetic analyses (7–19), while for fullerenol there is no evidence of available methods. Dox was analyzed by fluorescence labeling, radiolabeling, enzyme-linked immunosorbent assays, mass spectrometry, and chromatography in biological samples (7). Novel LC methods for Dox have been applied to biological and environmental samples (15), blood (18), blood cell lines (19), urine, and serum (20). On the other side, CE methods have been successfully developed for analyses of pharmaceutical final dosage forms (9, 12), plasma (14), environmental (15), and serum samples (17). The MEKC method has been employed for the analysis of neutral and ionic drugs in many research works in the past two decades. The most commonly MEKC-LIF (micellar electrokinetic capillary chromatography laser-induced fluorescence) method was used for Dox analysis in urine (8, 16) and cancer cell lines (11). On the other hand, the MEKC system with DAD was used for plasma (14) and cancer cell line samples (13). On the contrary, there are no chromatographic analyses so far reported for fullerenol C60(OH)24; fullerenol was mostly used as a matrix system in a few spectrometric methods (21–23).
Therefore, this paper covers the first-time development, optimization, and validation of MEKC for analysis of Dox and fullerenol in rat serum.
EXPERIMENTAL
In vivo testing
Fullerenol C60(OH)24 (98.8 % purity) was synthesized from polybromine derivative C60Br24 and characterized (Faculty of Science, Department of Chemistry, University of Novi Sad, Serbia) (4). To get 10 mg mL–1 fullerenol solution it was dissolved in a sterilized and apyrogenic NaCl (0.9 %):DMSO (80:20, m/ m) inside a laminar flow cabin immediately before use. Dox (Adriblastina®, doxorubicin hydrochloride lyophilizate for i.v. administration) was obtained from Pharmacia & Upjohn (Italy). The 2 mg mL–1 solution of Dox was obtained after dissolving in a sterilized and apyrogenic 0.9 % NaCl solution inside a laminar flow cabin immediately before use.
Male Wistar rats (Medical Experimental Centre, Ljubljana, Slovenia) were obtained at 7 weeks of age, quarantined, and housed 3–4 per cage at a 22–23 °C room temperature, 55 ± 10 % humidity and a 12-h light/dark cycle. They had free access to a standard laboratory diet (Altromin, Germany) and water. All experiments were approved by the National Animal Ethical Committee of the Republic of Slovenia (license number 34401-61/2007/7) and were conducted in accordance with the European Convention for ETS 123. The animals were randomly divided into six groups (10 per group) as follows:
1 – control healthy group (C) – rats received saline only ( i.p.)
2 – Dox group – i.p. Dox 1.5 mg kg–1
3 – Frl/Dox group – i.p Frl 100 mg kg–1 30 min before i.p. Dox 1.5 mg kg–1
4 – Frl/Dox group – i.p. Frl 50 mg kg–1 30 min before i.p. Dox 1.5 mg kg–1
5 – Frl/Dox group – i.p. Frl 25 mg kg–1 30 min before i.p. Dox 1.5 mg kg–1
6 – Frl group – i.p. Frl 100 mg kg–1.
For the purpose of this study a single dose was given to the animals and blood sampling was done after 2 h, 4 h, and 7 days. The blood for analysis was taken from the orbital venous sinus. Afterward, the animals were used for a long-term chronic toxicity study with the application of weekly doses for several weeks. Serum and several organs have been collected for oxidative stress testing (4, 5).
Samples
Dox and Frl stock solutions were prepared at a concentration of 500 mg L–1 in distilled water and were stored under two conditions (refrigerated at 4–8 °C and at room temperature 22–25 °C with a humidity of 40-65 %). Refrigerated solutions were used for all analyses. The stock solutions were diluted with distilled water to obtain concentrations ranging from 0.5 to 500.0 mg L–1.
One milliliter of rat serum was treated with 0.5 mL of precipitant (acetonitrile), centrifuged (5 min at 11000 rpm) and the supernatant was used for final electrophoretic analysis without dilution.
Capillary electrophoresis
An Agilent 3D-CE DAD capillary electrophoresis system (Germany) with HP ChemStation software for MECK analysis was used. The fused silica capillary (Agilent, Germany) with a length of 48.5 cm was selected as optimal (40 cm to detector × 50 µm id fused-silica capillary with bubble cell, 150 µm). The temperature and voltage were 25 °C and 25 kV, resp., while hydrodynamic injection was performed at 5.0 kPa for 100 s. Samples were measured at 234 nm.
The background electrolyte (BGE) was composed of 10 mmol L–1 borate (pH 9.3) plus 15 mmol L–1 phosphate (pH 7.0) buffer. Buffer solutions were prepared from NaH2PO4 or Na2B4O7 (Kemika d.d., Croatia) as aqueous solutions, and the final background electrolyte pH was adjusted to 7.0 with HCl. As surfactant, 15 mmol L–1 sodium dodecyl sulfate (SDS, Riedel-de Haën AG, Germany) was used with the addition of methanol (10 %, V/ V, Merck KGaA, Germany).
The capillary was conditioned before the first use by rinsing with 1 mol L–1 NaOH for 20 minutes, followed by 0.1 mol L–1 NaOH for 10 minutes and water for 10 minutes. The final step consisted of a wash with BGE for 20 min. At the beginning of each working day, the capillary was activated with 0.1 mol L–1 NaOH for 5 min under high pressure and then rinsed with water for 5 min and BGE for 10 min.
Method validation
Procedures and characteristics used for validation were those described in the literature (24–32).
Selectivity/specificity. – These parameters were preliminarily examined in model aqueous media. The method was also tested when control healthy sera were spiked with Dox and Frl and by monitoring the interfering peaks. The third set of fit-for-purpose experiments was conducted, with a slight deviation from the current ICH guidelines. Spiking of real samples of the 3rd investigated group (which received 100 mg kg–1 of Frl and 1.5 mg kg–1 of Dox), with both analytes at the maximal concentration at the linearity range (500 mg L–1 for each) was also performed.
Linearity. – Ten different concentrations of both components were used for linearity testing. Linearity was checked over the range of 0.5−500 mg L–1 using aq. standard solutions. The linearity of the analytical response of both investigated molecules was determined over the concentration ranges with a minimum determination coefficient of 0.99 ( R2). LOD (or DL) ( S/ N = 3) and LOQ (or QL) ( S/ N = 10) were determined using a baseline noise method. LLOQ and ULOQ were also determined. LLOQ and ULOQ are the lowest and the highest amount of an analyte that can be quantitatively determined with acceptable precision and accuracy, resp. According to ICH (30) and FDA guidelines (31) for LLOQ in chromatographic methods, the highest permissible inaccuracy is ± 20 %, and imprecision (as RSD) is 20 %.
Accuracy. – The accuracy of the method was controlled by analyzing a solution of known concentration (standard solution, spiked serum sample, and real samples) and comparing the measured and known concentration values. For both Dox and Frl tests have been done through 5 repetitions using 6 different concentrations within the linearity range (5, 10, 25, 50, 75, and 100 mg L–1).
Precision. – Model precision was measured within one day (intra-day) and inter-day (three different days) using standard solutions of Dox and Frl. Precision was measured at 25, 50, and 100 mg L–1.
Robustness. – For robustness testing the optimal MEKC conditions were slightly modified using one-variable-at-the-time and multi-variable-at-a-time approach including all possible parameters: electrolyte concentration (± 1 mmol L–1 each borate and phosphate buffer), buffer pH (± 0.1), SDS concentration (± 1 mmol L–1), capillary temperature (± 1 °C), applied voltage (± 0.1 kV), sample injection time (± 1 s), detection wavelength (± 3 nm), sample injection pressure (± 103.4 Pa), and capillary (different production batch) (27).
Sample stability. – To test the stability of both stock solutions two conditions have been applied (refrigerated 4 ± 2 °C and room temperature 23 ± 2 °C) over 30 days period (time-points: 0, 1, 2, 5, 10, 15, 20, 30 days). For every time point, 3 repetitive measurements have been performed.
Quality control standards. – QC standards have been prepared in accordance with ICH quality standards (25, 30), at 4 different concentrations. Blank serum samples from the control healthy group have been spiked with 0.5, 1.5, 250, and 400 mg L– 1 of Dox and 10, 30, 250, and 400 mg L– 1 of Frl. After every 10th analysis, QC samples have been tested. RSD values (n = 50) ranged from 2.2 – 3.7 and 3.9– 5.9 % for migration time and peak area, resp., for Dox; for Frl respective values were 4.5– 5.8 and 5.5– 8.2 %.
RESULTS AND DISCUSSION
Optimization tests
In a preliminary study a mixture containing Dox and Frl at 50 mg L–1 and 25 mg L–1, resp., was used. The influence of pH was examined over the range of 5.0−10.0 by using phosphate buffer and/or borate buffer in different ratios as electrolytes in deionized water and adjusting with HCl or NaOH to the required pH. The results showed that the best separation of doxorubicin and fullerenol was achieved with phosphate/borate buffer (pH 7.0) as BGE-containing SDS (MEKC system).
During the preliminary study peaks of both compounds showed shoulders and improper symmetry (tailing effect). Since the addition of different organic modifiers might be essential for the peak’s purity, methanol, ethanol, and acetonitrile were tried in concentrations up to 15 %. The presence of 10 % ( V/ V) methanol as an organic solvent in the BGE resulted in improved symmetry of peaks and shoulders disappeared.
The borate and phosphate buffer molarity varied from 5 to 50 mmol L–1 using the experimental conditions mentioned above in the experimental section. The final buffer was chosen to be the mixture of 10 mmol L–1 borate buffer pH 9.3 and 15 mmol L–1 phosphate buffer pH 7.0. The final background electrolyte pH was adjusted to 7.0 with HCl, and a suitable peak shape was achieved. The influence of higher SDS concentrations in the BGE on migration was minimal since in this case, peaks showed good symmetry and resolution without overlapping.
The results demonstrated that SDS concentration (10−100 mmol L–1) influenced the mobility of Dox and Frl. A concentration of 15 mmol L–1 SDS was selected for further experiments, giving the best shape and symmetry of both peaks and their resolution.
The effects of running voltage in the range of 5−30 kV were tested using a mixture buffer as stated above with 15 mmol L–1 SDS and 10 % ( V/ V) methanol, with final BGE pH 7.0, without running pressure, at 25 °C. The best results were obtained at 25 kV and an acceptable level of baseline noise was achieved by performing experiments at 25 °C.
The applied pressure was tested in the range of 0−5.0 kPa as well as injection time from 10 to 100 s, using the experimental conditions given above. Migration times decreased with increasing the applied pressure. A pressure of 5 kPa can be selected as optimum with 100 s of injection time. It gives the best peak symmetry and acceptable level of baseline noise.
Electropherograms for aq. standards and real samples under selected conditions are presented in Figs. 1 and 2. At 234 nm, Dox and Frl were detected at 6.8 and 11.5 min in aq. standard solutions, whereas in rat serum after administration tRs shifted to 7.2 and 12.9 min, respectively.
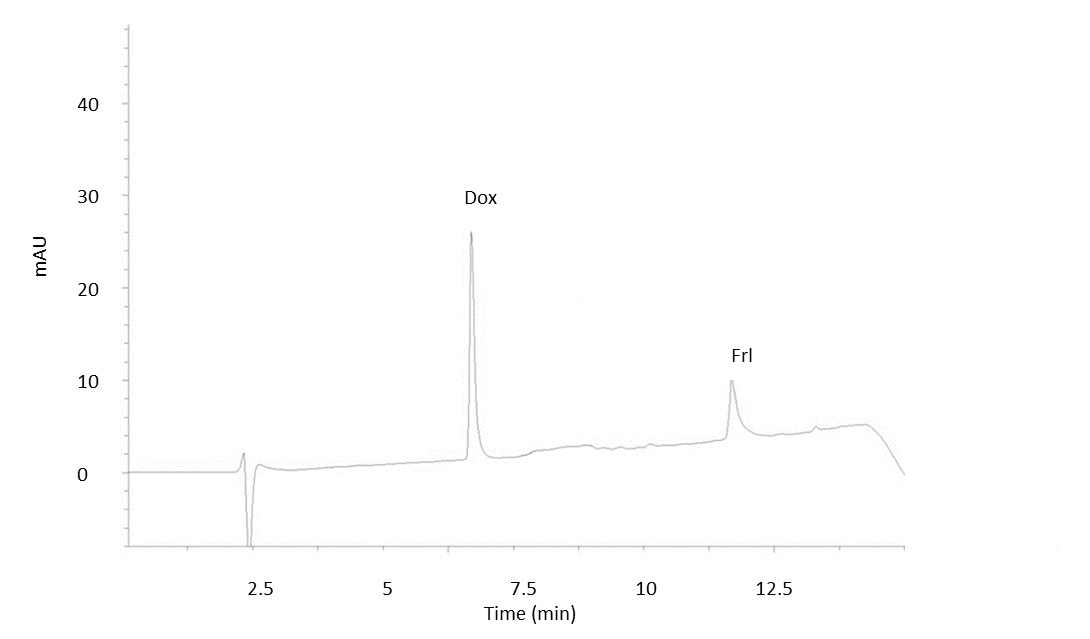
Fig. 1. MEKC electropherogram of doxorubicin aqueous standard (50 mg L–1) and fullerenol aqueous standard (25 mg L–1) under optimal separation conditions (see Experimental).
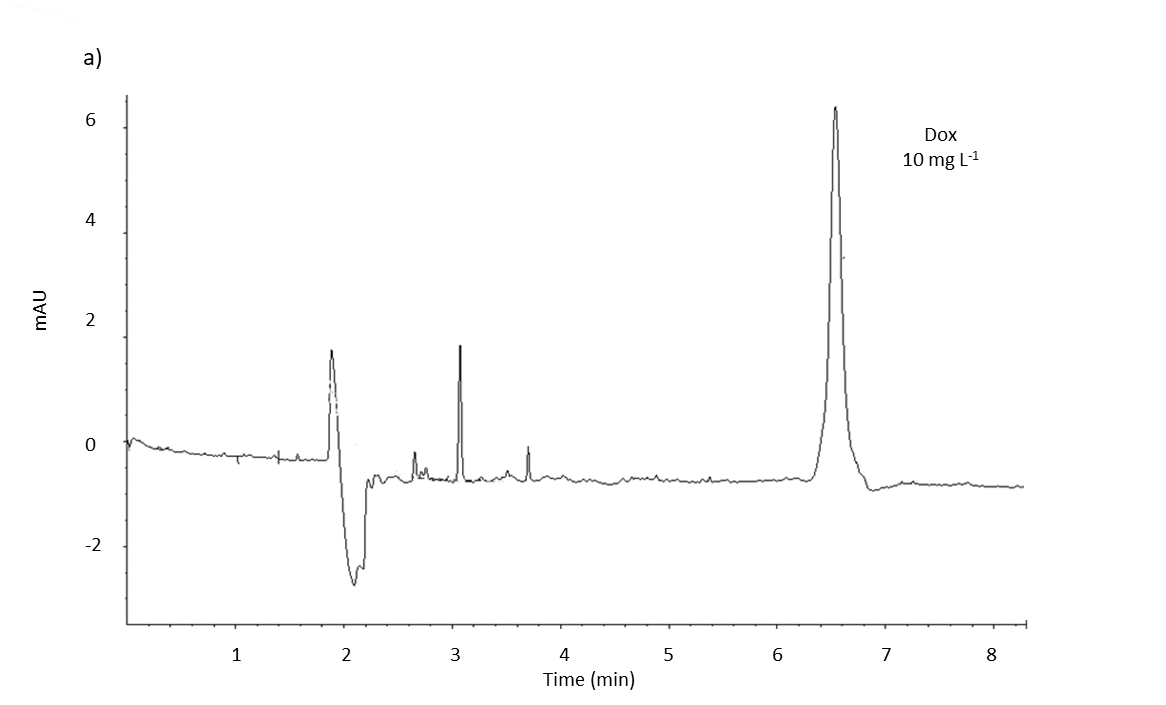
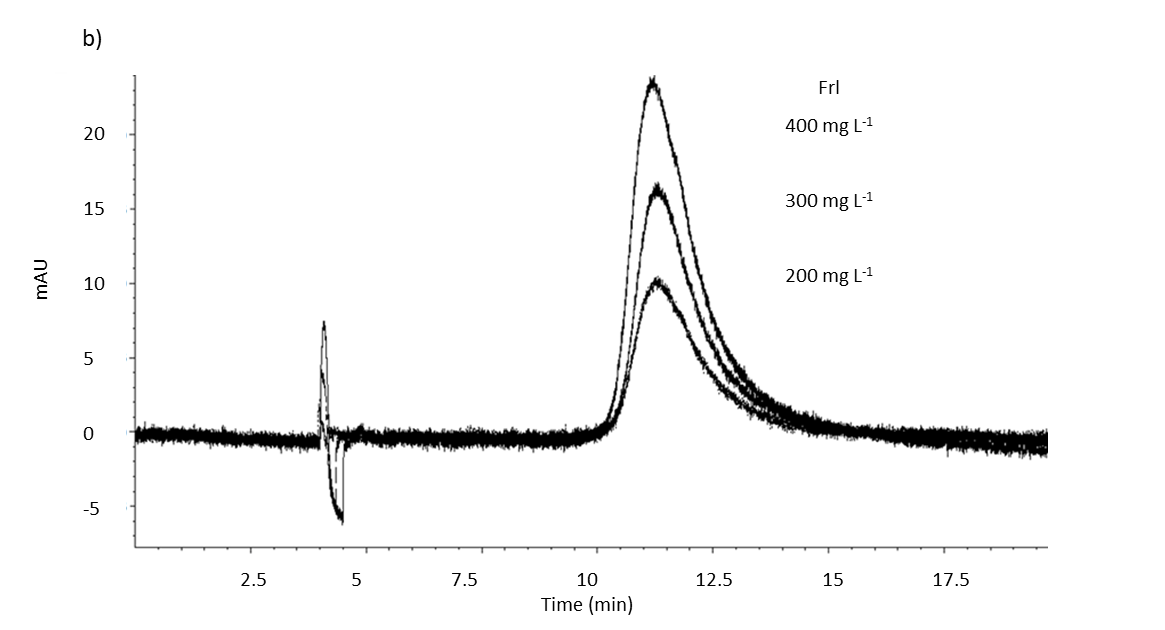
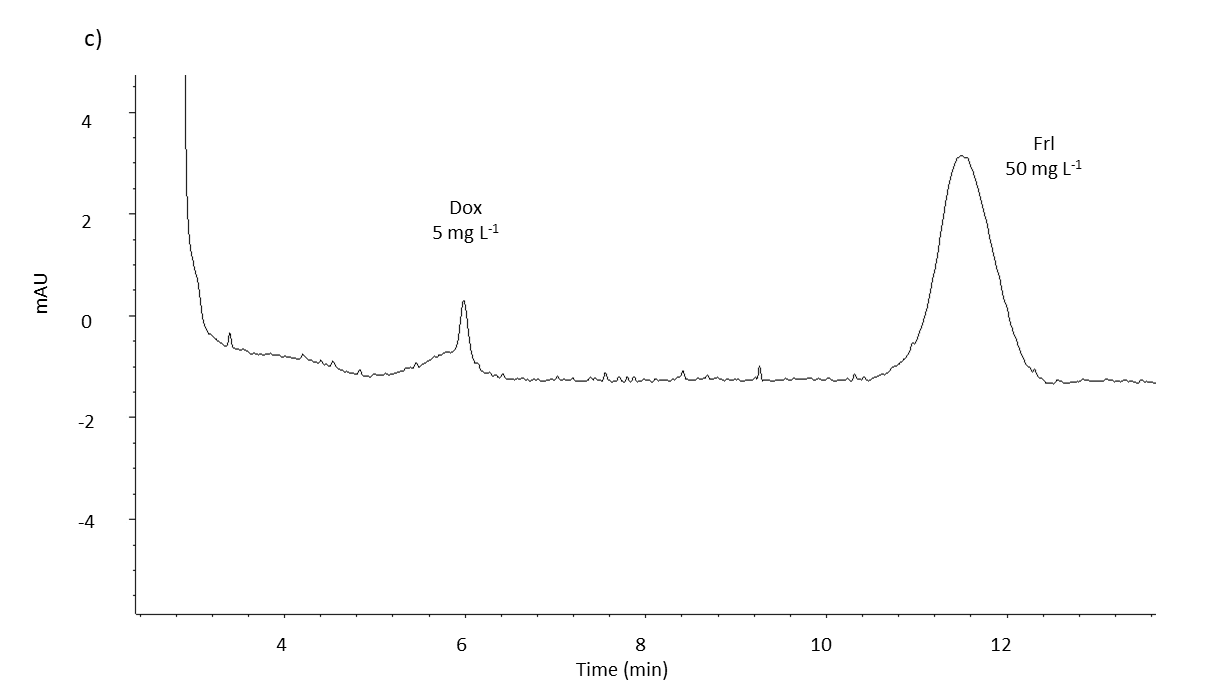
Fig. 2. MEKC analysis electropherogram of spiked blank serum with: a) doxorubicin standard (10 mg L–1); b) fullerenol standard (at three different concentrations: 200, 300 and 400 mg L–1), and c) doxorubicin (5 mg L–1) and fullerenol (50 mg L–1) standards, under optimal separation conditions (see Experimental). Blank serum was used from the control group (group 1).
In all previous papers dealing with Dox, separation was achieved very fast and below 10 min (8–20) which is in good alignment with our MECK method. However, due to the complexity of the Frl structure and size, and the need to achieve good separation, its peak migration time is a bit longer, up to 12 min. Using more aggressive conditions such as higher SDS concentration, higher pressure, higher voltage, longer injection time, or higher amount of organic modifier, would result in significant deformation of Frl peak during separation as well as overlapping with Dox.
Chemical basis – discussion
Dox (usually in the form of hydrochloride salt) is a small molecule (diameter 1.5 nm, Mr 543), it is a weak base and is quite stable in water when refrigerated (1). Most of the previous analytical papers published the separation of Dox from other anthracyclines and antibiotics in different samples and almost always SDS was used as a surfactant and BGE buffer with pH between 9.0 and 9.6 (9, 12, 14). Due to the high number of tested compounds and their different characteristics, in several studies’ a higher concentration of SDS was used (9, 11, 12, 14) compared to 15 mmol L– 1 which we found optimal in this study. In two cases (8, 16), SDS was used bellow the critical micellar concentration at room temperature, namely at 10 mmol L– 1 . However, in a review by Flurer (16), there is an example with 10 mmol L– 1 SDS, while on the other side Yu et al. (8) used only 5 mmol L– 1 of SDS. The major reason for using 15 mmol L– 1 SDS in our case is due to characteristics of Frl (5) ( Mr 1128, diameter 1 nm). The challenge of Frl is in its spheric structure and electron distribution within the spheric cage. The acidity of Frl mainly starts from the H+ donation mechanisms and the reducibility mainly from the H˙ donation mechanisms (29). Both of those reflect the extent of stabilization of C60-cores' π-electron configurations accompanying the H+ and H˙ donations. High acidity and reducibility of Frl is a consequence of a large π-electron stabilization effect associated with the H+ and H˙ donation reactions, in both directions, which means that Frl is pH sensitive compound. Frl with stable π-electron configuration of C60 core has a low acidity and reducibility, and vice versa. This makes a strong dependence on acidity and reducibility of Frl on its hydroxyl distributions. This was driving us to optimize the method as close to neutral pH as possible. Finally, for both molecules, pH 7.0 was acceptable for separation.
With regards to spectrum, it is well documented in the literature that Dox has several high absorption peaks between 215 and 530 nm (9); mostly used for detection were at 215, 230, or 254 nm (9, 12, 16). At the same time spectrum of Frl has the highest peaks at 210, 234, and 290 nm. During preliminary tests two were selected: 215 and 234 nm, but finally, the best symmetry of peaks and separation was recorded at 234 nm.
Method validation results
Sensitivity and linear regression data, recovery, and precision for the investigated MEKC method are presented in Table I.
Table I. Sensitivity, accuracy, and precision of the MEKC method
a Mean ± SD, n = 10;b Serum from each animal group has been spiked with 50 mg L–1 doxorubicin and 25 mg L–1 fullerenol.
With regards to Dox, there are several review and original papers published recently dealing with quality control of pharmaceutical forms (9, 11, 12), or biological samples (8, 14, 17). In all cases, a common suggestion for Dox separation and analysis was the usability of the micelle (8, 11, 12, 14, 17). Xiao et al. (8) performed micellar electromagnetic chromatography using 10 mmol L–1 borate buffer containing 20 mmol L–1 Triton X-100, 5 mmol L–1 sodium dodecyl sulfate, and 30 % ( V/ V) methanol at pH 9.00. Although SDS was below minimal effective micellar concentration (10 mmol L–1) it made a strong micellar system in combination with Triton X-100, which was successfully used for the analysis of Dox in urine at high pH. In that investigation curcumin was tested together with Dox to avoid complex fluorescence derivatization and introduction of interfering components; a highly sensitive double wavelength excitation source LIF (D-W-Ex-LIF) detector composed of a 445-nm and 488-nm commercial laser diode was constructed for simultaneous detect. There, LOQ for Dox was almost 30 times lower (0.0100 mg L–1) than in our study (0.317 mg L–1), while our recovery was much better (100.9 ± 2.5 %) using the MEKC method in comparison to their separation conditions (94.9–109.1 %). It is also clear that our method is more simple and faster.
Ho et al. (11) developed a modified MEKC–LIF (micellar electrokinetic chromatography-laser-induced fluorescence) method suitable for analyzing Dox in biological samples. The MEKC migration buffer was made of 10 mmol L–1 borate, and 100 mmol L–1 SDS (pH 9.3). It was found to provide an efficient and stable electrophoretic separation and analysis for Dox in biological samples. Responses were linear in the range of 11.3 to 725.0 µg L–1. LOQ was found to be 43.1 µg L–1 and LOD as 6.36 µg L–1. This method was very sensitive and able to detect Dox in much lower concentrations than in our current work. However, the conditions of that study were not suitable for Frl, and especially high concentration of SDS had a negative influence on Frl peak shape as well as its retention time. We found overlapping of Dox and Frl peaks with higher amounts of SDS in BGE.
A very simple and fast (8 minutes) CE method for the separation of several anticancer drugs was presented by Guichard et al. (12). A capillary zone electrophoresis (CZE) method coupled to UV was developed with a BGE made of a 100 mmol L–1 phosphate buffer at pH 2.5 containing 50 % ( V/ V) of acetonitrile and dynamic coating of capillaries with CeofixR. This method allowed the analysis of doxorubicin with 9 other anticancer drugs in less than 8 min. The results presented in this study are comparable to our results. However, at pH 2.5 and without micelle, Frl separation would not be possible under these conditions.
Shakalisava and Regan (14) in their investigation developed CE, MEKC, and MEEKC methods for the separation of anthracyclines and taxanes. They concluded that for best separation micelles are recommended and both MEKC and MEEKC methods can be applied. An alkaline buffer, 25 mmol L–1 Tris-phosphate at pH 9, with high SDS (100 mmol L–1) and methanol (70 %), was used in the MEKC separation as it created a strong EOF for the negatively charged micelles to be swept towards the detector. A 100 mmol L–1 SDS resulted in sufficient resolution of investigated peaks and the EOF. Once again, under these conditions with high SDS and methanol concentration, Frl would not be sufficiently separated and detected in our case. Overall, we can see that Dox can be tested successfully at lower pH (2.5) as well as at higher pH (9.0–9.3). Best separations and detections have been demonstrated using micelle and the most often used surfactant was SDS. Both phosphate and borate buffers have been presented in recently published papers as acceptable for BGE, while methanol and acetonitrile in different amounts can be useful modifiers. Consequently, all of this was a good basis for the development of our method which is presented in this paper, however, modifications that have been made, are driven mainly by characteristics of Frl and its behavior in the MEKC system.
Our method demonstrated acceptable results related to investigated analytical performances, in standard solutions and in rat serum samples. In the case of real samples, migration time for both analytes shifted markedly (see Figs. 1–3 and Table II). This did not influence the peak shape or the peak area. We assume that strong EOF and/or some interactions with serum components (no additional extraction steps used) have been pushing both analytes within the capillary resulting in longer migration times. The formation of drugs' metabolites which still need to be identified may not be ruled out. Due to the fact that final results have not been impaired, we did not investigate this effect on retention time further. Anyhow, in more complex samples with lower doses of Frl, this phenomenon has to be investigated and clarified. Delay in retention time was observed in all real samples, no matter if they contained Dox alone, Frl alone, or both components.
Table II. Concentration of Dox and/or Frl in serum from Wistar rats at different time points
Bold numbers indicate measured compounds. C – control group (saline); groups 2–6: Dox alone (1.5 mg kg – 1 ), or Frl alone (100 mg kg – 1 ), or Dox and Frl together with different doses of Frl administered (25, 50 and 100 mg kg –– 1 ) + Dox (1.5 mg kg – 1 ).
a Mean ± SD, n = 10;b See Fig. 3.
Robustness testing was performed in aqueous medium. Results of robustness (one-variable-at-the-time and multi-variable-at-the-time) for three levels per parameter (minus one unit, at experimental conditions and plus one unit) are expressed as RSD values. Robustness testing data showed RSDs for tR and AUC ranging from 0.1 to 2.9 % for both analytes, including different capillary lots. Even more, simultaneous change of all parameters caused variability of 4.2–4.7 % for migration time and 7.6–8.9 % for AUC, including both directions of the change.
Regarding sample stability, for both analytes, the assay was above 98 % up to day 15. However, after day 20 Dox concentration at the room temperature dropped to 94.2 % and for Frl to 96.8 % but only after day 30. Both drugs remained at above 98 % when refrigerated samples (4 ± 2 °C) for up to 30 days. Therefore, it is recommended to keep both stock solutions under refrigerated conditions not longer than 30 days (Table III).
Table III. Stock solution stability testing of Dox and Frl under refrigerated and room conditions over 30 day period
a Accuracy, mean ± SD (n = 3).
The summary of MEKC parameters related to both standard and real samples are summarized in Table IV. It can be seen that the vast majority of the evaluated MEKC parameters are suitable. Somewhat lower number of theoretical plates can be observed, however, the resolution between the peaks is more than satisfactory.
Table IV. Key MEKC parameters overview for both standard and real samplesa
a Mean ± SD;bn = 10;c n = 90 (samples with Dox and Frl from groups 3, 4, and 5); 10 animal samples per group with 3 repetitions.
Pharmacology
Biological samples (serum) from 60 rats have been collected 2 h, 4 h, and 7 days after i.p. administration of both molecules. The control group was treated with saline, while other groups had Dox alone (1.5 mg kg–1), or Frl alone (100 mg kg–-1) or Dox and Frl together with different doses of Frl administered (25, 50, and 100 mg kg–1). Table II shows results for real samples, whereas Figs. 2 and 3 show electropherograms for serum samples recorded after spiking blank serum (Figs. 2a–c) with Dox or Frl, or administration of Dox and Frl together (Fig. 3). Due to limited absorption from the intraperitoneal area after administration of the Frl, final serum concentration is the same no matter of the dose. In previous studies, we have been confirming this effect and suggesting 25 mg kg–1 as a sufficient dose without significant segregation of Frl particles around the liver and intestine (4). This theory needs additional investigation using lower concentrations of Frl (equal and below 25 mg kg–1) and proper pharmacokinetic study.
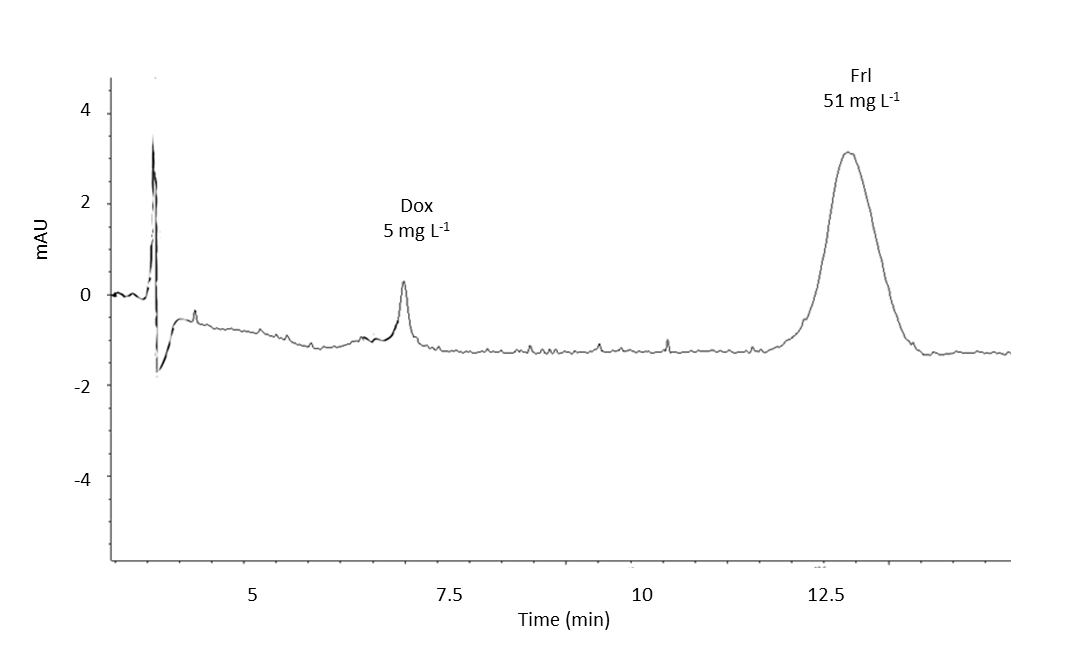
Fig. 3. MEKC analysis electropherogram of doxorubicin (4 h after administration) and fullerenol (4.5 h after administration) in Wistar rat serum under optimal separation conditions (group 3).
In accordance with existing pharmacokinetic results, Dox elimination is relatively fast (33) whereas Frl data are not available in the literature. The time points that we used in this study are not sufficient to support clear conclusions about basic pharmacokinetic parameters, therefore further investigations would be needed including many time points for Dox between time 0 and 48 h (such as 5 min, 15 min, 30 min, 1 h, 2 h, 4 h, 8 h, etc., up to 48 h). In the case of Frl due to slow elimination proven in this study, additional longer time points would be needed (such as 24 h, 48 h, 72 h, 5 days, 7 days, 10 days, 14 days, 21 days, up to 28 days).
CONCLUSIONS
The new MEKC method is proposed for rapid determination of Dox and/or Frl in biological samples without extraction, using SS as surfactant and BGE consisting of 10 mmol L–1 borate buffer pH 9.3, and 15 mmol L–-1 phosphate buffer pH 7.0 (with final pH 7.0 adjusted with HCl), with 10 % ( V/ V) methanol as the organic modifier. The presented method is simple, without complicated extraction procedures and/or internal standard use. However, in other in vivo models (pigs, chimpanzees, humans), it might be modified. The method demonstrated suitable selectiveness, precision, accuracy, and robustness. After validation, this method was successfully applied to the analysis of Wistar rats’ serum after administration of Dox and/or Frl. Its application can be extended for pharmacokinetics purposes, in combined therapy with Dox and Frl, or for Frl only. The next step would be modification of the method using other mammal serum samples (such as pigs) and full bioanalytical method validation, with extended pharmacokinetic evaluation.
Data availability. – The data that support the findings of this study are available from the corresponding author upon reasonable request.
Conflicts of interest.– None declared.
Funding– This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
Authors contributions. – Conceptualization, R.I.; methodology, R.I.; analysis R.I., M.S. and N.K.G; writing, original draft preparation, R.I.; writing, review and editing, R.I., M.S. and N.K.G. All authors have read and agreed to the published version of the manuscript.