
INTRODUCTION
A plethora of evidence indicates the involvement of fibroblast growth factor - fibroblast growth factor receptor (FGF-FGFR) – signaling in regulating multiple physiological processes, including angiogenesis, wound healing, and tissue regeneration (1). Dysregulation of this signaling pathway has been linked with tumorigenesis, tumor progression, and drug resistance to anticancer treatment in varieties of cancer (2). FGFRs are single-pass trans-membrane proteins in the receptor tyrosine kinases (RTK) family. Notably, the FGFR family has five members, namely, FGFR1, FGFR2, FGFR3, FGFR4 and FGFR5. Because of frequently observed aberration in the FGF-FGFR axis in cancers, FGFR targeting has received considerable attention as a potential therapeutic target. Four members of the FGFR family, i.e., FGFR1-4, have emerged as potential targets in different cancers (3).
This review aims to provide clinical data on the FDA-approved selective FGFR tyrosine kinase inhibitors (TKIs) for treating various malignancies, emphasizing the mechanism of action, pharmacological activities, toxicity, and resistance mechanisms. Additionally, we will briefly summarize ongoing investigations for unapproved therapeutic indications in clinical trials.
FGF-FGFR pathway
The mammalian FGF family comprises 23 proteins (FGF1–FGF23), classified as intracellular and secreted FGFs. The intracellular FGFs (FGF11–14) proteins act as cofactors for voltage-gated sodium channels and do not bind to cell surface receptors, whereas secreted FGFs act extra-cellularly by binding to FGFRs to initiate signal transduction (4). Secreted FGFs are further categorized as endocrine FGFs, including FGF15/19, 21, and 23, and canonical FGFs, such as FGF1-10, 6-18, 20 and 22. Both types of FGFs exhibit their biological effects by binding with FGFRs on target cell membranes (5). Still, they differ in the distance before their binding to FGFRs. While canonical FGFs interact as autocrine or paracrine factors, with heparan sulfate proteoglycans (HSPGs) as cofactors, endocrine FGFs travel a considerable distance in blood circulation before binding FGFRs with the help of the Klotho protein family, comprising αKlotho, βKlotho, and γKlotho (3, 5–8). The canonical FGFs and FGFRs interaction mediates cellular survival, metabolism, proliferation, tissue metabolism, organogenesis, inflammatory responses, and repair. In addition to the canonical FGFs’ action, endocrine FGFs contribute to bile acids, lipids, and glucose metabolisms (3, 5, 9). Deregulation of FGF's expression, secretion, and degradation may influence multiple biological activities such as metabolism, organogenesis, and wound healing, and may even lead to various cancers (10).
The FGFR family includes four protein kinases (FGFR1–4) and a non-protein kinase, FGFR5 or FGFRL1 (11). These receptors differ in ligand-binding selectivity and tissue distribution (12). Similarly, alternative splicing is the source of heterogeneity and ligand specificity for the four receptors involving splicing into 48 different isoforms. The FGFR protein comprises three external immunoglobulin-like domains (IgI–III), a transmembrane helix, and an intracellular kinase domain. An acid box composed of a short stretch of aspartates and glutamates is present between IgI and IgII, which exhibits auto-inhibitory function along with the Ig-I domain in the absence of FGF-binding, whereas, IgII and IgIII domains are required for ligand binding. The FGFR1–3's ligand selectivity is regulated by alternatively spliced IgIIIb/c loop variants of the receptors with different biological consequences, predominantly expressed in epithelial and mesenchymal cells, resp. (13, 14). The encoded IgIII domains in FGFR4 do not undergo alternative splicing. The tissue-specific expression of the IgIIIb/c variants governs the interaction in cancer, embryogenesis, tissue repair, and maintenance (15–17).
The FGF binding to FGFR results in dimerization, transphosphorylation, and activation of the tyrosine kinase domain, which subsequently activates several downstream signaling pathways (18). Activated FGFRs require various proteins to activate downstream signaling pathways, such as FRFR substrate 2 protein (FRS2) and other interacting proteins, including GAB1, GRB2, SOS, and PLC-γ. Various downstream signaling mechanisms are involved in the FGF-FGFR axis associated with multiple physiological functions. Fig. 1 presents the PLC-γ/PKC, PI3K/AKT/mTOR, PI3K/AKT/MAPK, RAS/RAF/MEK/ERK and JAK-STAT pathways as a flow chart (19–23). Regarding downstream signaling, the four receptors (FGFR1–4) differ from one another due to variances in their affinities and specificities for intracellular adaptor proteins, subcellular transport patterns after receptor activation, and rates of receptor endocytosis. The activation of downstream pathways shows similarities between FGFR1 and FGFR2. Nevertheless, significant differences can be observed when compared to FGFR3 and FGFR4 (12). Depending on the type of FGFs and the receptor, the FGF-FGFR pathway is engaged in multiple physiological processes. FGFR1 is crucial for embryo development, cell cycle regulation, wound healing, and phosphorus homeostasis. The main functions of FGFR2-3 are blood vessel creation, wound healing, cell cycle regulation, and embryonic development. Although FGFR4 is implicated in tissue repair, embryogenesis, and angiogenesis, it has substrate-specific effects on glucose homeostasis, bile acid metabolism, and vitamin D homeostasis (10, 24).
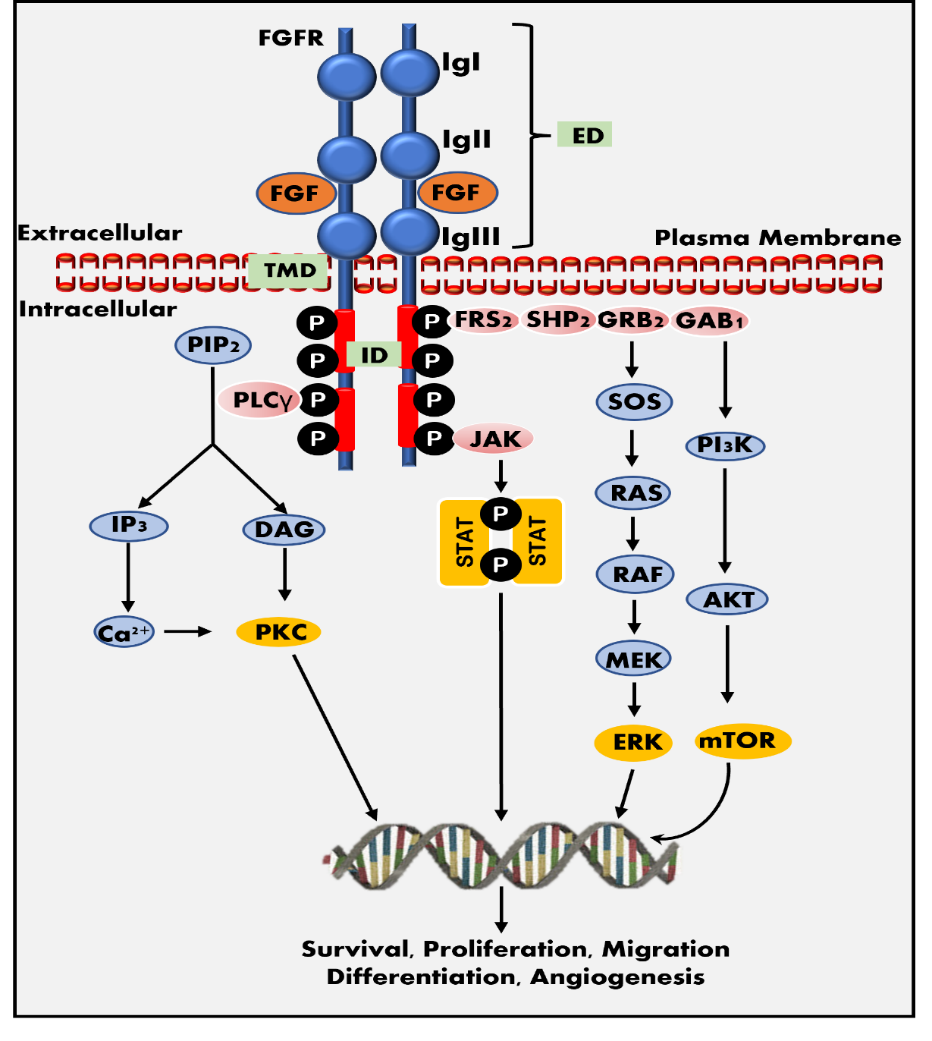
Fig. 1. FGF-FGFR signaling pathway. FGFR structure: the receptor contains three extracellular immunoglobulin-like domains (ED): IgI, IgII, and IgIII, an intracellular tyrosine kinase domain (ID), and a trans-membrane domain (TMD). The IgI and IgII domains comprise the FGF binding site. FGFR pathway: the binding of FGF to FGFR tyrosine kinase domain may activate several signaling pathways required for cellular survival, proliferation, migration, differentiation, and angiogenesis, including the JAK-STAT pathway, the PI3K-AKT-mTOR pathway, and the RAS-RAF-MEK-ERK pathways.
Therapeutic potential of FGFR tyrosine kinase inhibition
The FGFR tyrosine kinase family is the major driving factor for carcinogenesis mediated by FGF overexpression, FGFR genetic alterations, angiogenesis, and immune tolerance in the tumor microenvironment (25). A next-generation sequencing investigation of 4,853 solid tumors revealed FGFR abnormalities in 7.1 % of malignancies. Among the reported FGFR aberrations, gene amplification accounted for the major contribution towards malignancies, followed by gene mutations and gene rearrangements. Receptor-wise, the most changes were found in FGFR1 (49 %), followed by FGFR3 (23 %), FGFR2 (19 %), and FGFR4 (7 %) (26).
Another next-generation sequencing analysis of 5557 Chinese patients with solid tumors found FGFR1–4 mutations in 9.2 % of cases, with almost 50 % of cases, gene amplifications, and mutations were the most frequent, and gene rearrangements (10.0 %) were the least frequent (27). Of the 5557 cases studied, 4.6 % were associated with FGFR1, 2.1 % with FGFR2, 1.6 % with FGFR3 and 1.4 % with FGFR4. FGFR1-4 mutant malignancies were seen in almost all types of solid tumors, with breast cancer, endometrial carcinoma, gastric cancer, and sarcoma being the most common types. Other atypical anatomical sites include skin, reproductive organs, bone, digestive, nervous, and hematological systems harboring FGFR1–3 gene alterations (25).
With the growing understanding of FGFR-driven cancer therapeutics, FGFR inhibition has emerged as a potential targeted strategy. Various FGF ligand traps and monoclonal antibodies have been developed to target FGFR on extracellular receptor domains. Multiple small molecule inhibitors targeting FGFR intracellular tyrosine kinase domain are also explored. These small-molecule inhibitors can be FGFR selective or non-selective, inhibiting other tyrosine kinases such as PDGF and VEGF (3, 10, 28). Even though kinase inhibitors were introduced as a cancer treatment in the twentieth century, today, a multitude of kinase inhibitors are being studied in clinical or preclinical research to specifically target FGFR-altered carcinomas and to overcome the drug-resistance in chemotherapy and other targeted therapies (29, 30). A recent study has shown that FGFR inhibitors have the potential to be used off-label in a wide range of cancer types and patient populations (31). Based on population cancer statistics and the frequency of molecular alterations, off-label use may be three times more common than on-label use. In the following section classification of small-molecule FGFR-TKIs is discussed.
Classification of FGFR inhibitors
Promising results have been documented with FGFR inhibitors as a drug class, which target FGFR receptor abnormalities in various cancers (32) . Small molecule FGFR-TKIs may be classified into two generations. The first-generation TKIs include ponatinib, lucitanib, nintedanib, dovitinib, derazantinib, and anlotinib, which act as non-selective TKIs of multiple tyrosine kinases, including FGFR, BCR-ABL, VEGFR, c-Kit, PDGFR and FLT-3 (3, 33). Due to the conservation of the hinge region and activation loops around the ATP-binding site in FGFR-related receptor tyrosine kinases, most first-generation TKIs were multi-kinase FGFR inhibitors (25). On the other hand, second-generation TKIs have selective inhibitory activity against FGFRs. Based on target specificities, selective FGFR TKIs may be categorized as FGFR1-3 inhibitors, FGFR4 inhibitors, dual kinase inhibitors, and pan-FGFR inhibitors ( Fig. 2) (25).
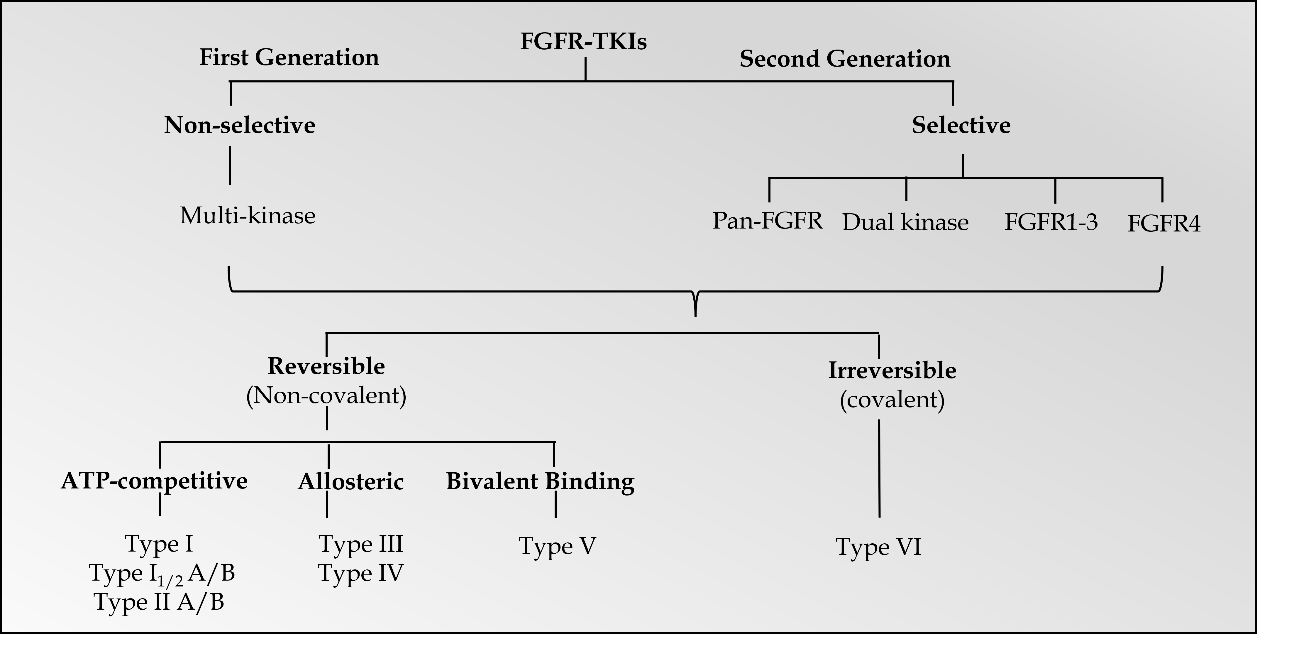
Fig. 2. Classification of FGFR-TKIs.
Further categorization of FGFR TKIs includes reversible and irreversible inhibitors (34, 35). A recent review divided the small molecule protein kinase inhibitors into seven main groups: type I, I1/2, II, III, IV, V, and VI inhibitors (36). The first six types were categorized as reversible or non-covalent inhibitors, and type VI inhibitors bind irreversibly to the targeted receptors via covalent bonding. Among categorized groups, types I, I1/2, and II inhibitors exhibit binding in and around the ATP binding pockets of the receptors. Type I and 11/2 inhibitors bind to FGFR's ATP-binding domain in the active DFG-in configuration, and type II inhibitors bind to FGFR's ATP-binding domain in the inactive DFG-out configuration. For instance, AZD4547, a type I inhibitor, binds to FGFRs in the active DFG-in configuration, whereas ponatinib, a type II inhibitor, binds to the inactive DFG-out configuration of FGFRs and other RTKs (37, 38). Furthermore, types I1/2 and II antagonists are further divided into A and B subtypes based on whether binding extends to hydrophobic back cleft (A) or not (B).
Type III and IV inhibitors demonstrate allosteric binding either next to the ATP-binding site or at a distance away from the ATP-binding site (39). Allosteric FGFR inhibitors are small molecule inhibitors that impede the signaling pathway by attaching to a place other than the ATP-binding site in FGFRs. Alofanib (RPT835) is a representative of this class of inhibitors. Alofanib inhibits FGF2-induced phosphorylation of FRS2 in FGFR2-expressing cancer cells by binding selectively to the extracellular IgIII subdomain of FGFR2 in the nanomolar range. SSR128129E is another orally active, allosteric FGFR1 inhibitor that promotes crucial conformational changes in the FGFR1 IgIII subdomain, resulting in faulty FGFR internalization (40). Type V inhibitors are bivalent inhibitors spanning two kinase domain regions of the targeted kinases. Moreover, antagonists that form covalent bonds with their targets were categorized as type VI inhibitors (29). Covalent (irreversible) inhibitors are thought to have increased binding affinity and selectivity. Futibatinib, BLU-554, and FGF401 are irreversible covalent inhibitors of FGFRs (30).
FDA-APPROVED SELECTIVE FGFR INHIBITORS
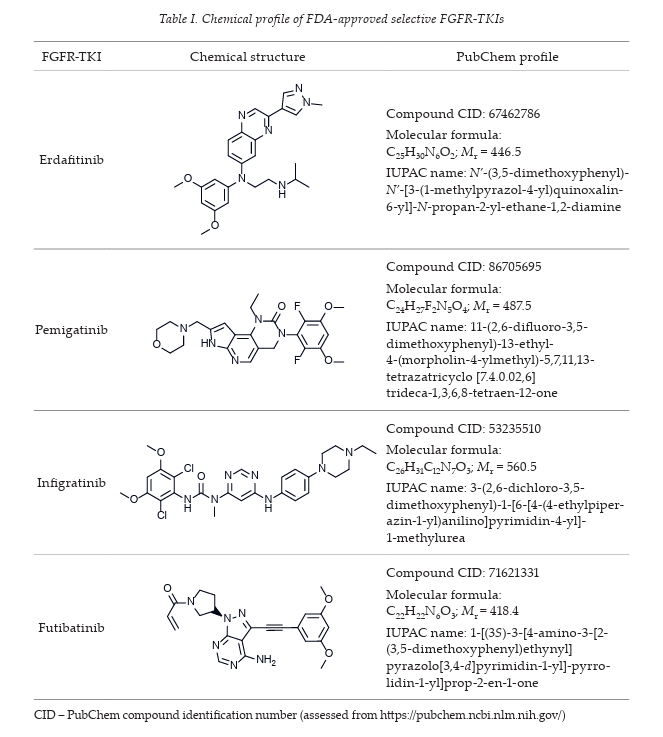
Several FGFR-TKIs have been developed; the FDA has approved four selective FGFR TKIs, including erdafitinib, pemigatinib, infigratinib and futibatinib (chemical profile summarized in Table I), as a treatment against oncogenic FGFR-driven cancers such as urothelial carcinoma, intrahepatic cholangiocarcinoma, and relapsed or refractory myeloid/lymphoid neoplasms.
Erdafitinib
Erdafitinib, also known as JNJ-47856493, first developed by Janssen Pharmaceutical Co. in collaboration with Astex Pharmaceuticals, is an orally active, small, potent pan-FGFR tyrosine kinase inhibitor (41). Erdafitinib is a quinoxaline derivative, potently inhibiting FGFR1-4 kinases with IC50 values in the range of 1.2–5.7 nmol L–1. Erdafitinib binds to the FGFR kinase in an inactive DFG-in conformation via the formation of hydrogen bonds with Ala564 and Asp641 residues in the hinge region and the DFG motif, resp. The molecular interactions of erdafitinib with FGFR are presented (Fig. 3a) according to the co-crystallized complex retrieved from the server of the protein data bank (PDB) bearing the ID 5EW8. A favourable intermolecular hydrogen bonding interaction was noted between the nitrogen atom of the quinoxaline core and the H-N group of the aliphatic amino acid Ala564. Heteroatoms of the drug structure binding specifically to this amino acid alanine (Ala564) are very crucial for the inhibition of the functional aspects of FGFR. Thus, this hydrogen bonding interaction is considered to be significant along with other interactions. The pyrazolyl moiety in erdafitinib was surrounded by simple aliphatic amino acid Gly567, while, the aliphatic side chain showed hydrophobic interactions with Leu630. Further, dimethoxy substituted aromatic ring in erdafitinib exhibited hydrophobic aromatic interactions with the amino acid residues such as Asn628, Ala640, Val492, and Leu484. Erdafitinib demonstrates modest off-target effects against other kinases, such as BLK, RET, ABL1, LYN, LCK, TIE1, KIT, PDGFRA-B, and VEGFR2. Similarly, in a cell assay against various cancer cell lines with FGFR genetic aberrations, erdafitinib exhibited potent inhibitory potentials with nanomolar IC50 values (42, 43). A preclinical study also investigated the anticancer and radiosensitizing potentials of erdafitinib in colorectal cancer (44). In the results, erdafitinib effectively suppresses cell proliferation and cell survival in vitro and in vivo in NCl-H716 cell lines with the highest expression of FGFR2. However, erdafitinib showed no radio-sensitizing effect on NCl-H716 cells either in vitro or in vivo.
Based on the findings from preclinical studies, erdafitinib was studied in an open-label phase II trial (NCT02365597) to assess the safety and efficacy in patients with metastatic or surgically unresectable urothelial cancers harboring specific FGFR genomic alterations (45). A total of 99 patients received an average of five cycles of erdafitinib [8 mg QD continuously, 9 mg QD as bioactivity-guided dose escalation (QD – quaque die, namely, once-a-day)]. Using the erdafitinib was accompanied by a confirmed response rate of 40 % in patients who had had chemotherapy or neoadjuvant/adjuvant chemotherapy in the past, 3 % of patients had a complete response and 37 % had a partial tumor response rate. Treatment-related grade 3 or higher adverse events, manageable by dose adjustments, were recorded in nearly half the patients, with zero mortality. The most commonly reported adverse effects were hyperphosphatemia (72 %), skin changes (48 %), nail changes (51 %) and eye disorders (51 %).
Similarly, 24 months longer follow-up study was carried out, including 101 patients at 126 medical centers in 14 different countries (46). The findings showed a safety profile similar to the initial study; 72 % of patients experienced treatment-related adverse events in grades 3-4. The most common adverse events were stomatitis (14 %) and hyponatremia (11 %) (47).
An exploratory association between serum phosphate levels and efficacy or safety outcomes was examined in a different investigation (48). The Cox proportional hazard or logistic regression models used in the exposure-response analyses on 177 patients provided evidence in support of the notion that the incidences of specific adverse events related to erdafitinib treatment increase with higher serum phosphate levels and that activity-guided dose titration may be used to optimize erdafitinib's therapeutic benefit-risk ratio. On March 15, 2018, the FDA designated erdafitinib as a breakthrough treatment for urothelial carcinoma, the sixth most prevalent type of cancer in the USA, premised on impressive outcomes from this clinical trial.
Erdafitinib was subsequently given expedited FDA approval on April 12, 2019, to treat adult patients with locally advanced or metastatic urothelial cancer who had progressed during or after at least one line of prior platinum-containing chemotherapy, and had specific FGFR3 mutations or FGFR2 or FGFR3 fusion. The FDA-approved companion diagnostic test (therascreen FGFR RGQ RT-PCR kit; QIAGEN GmbH - Germany.) should be used to select the patient for erdafitinib treatment (49). A comparative phase III study is recruiting patients with advanced urothelial cancer and FGFR alterations to compare erdafitinib with the standard of care treatment consisting of chemotherapy (docetaxel or vinflunine) or anti-programmed death ligand-1 (PD-L1) agent pembrolizumab (NCT03390504).
Eventually, erdafitinib emerged as the first FDA-authorized selective FGFR tyrosine kinase inhibitor and is now being explored as a therapeutic option for liver cancer, cholangiocarcinoma, non-small cell lung cancer, lymphoma, prostate cancer, breast cancer, and esophageal cancer. Erdafitinib was administered in ascending doses ranging from 0.5–12 mg QD continuously or 10–12 mg intermittently (7 days-on/off) to patients with advanced solid tumors in a phase I dose escalation trial (NCT01703481). The recommended phase II dose (RP2D) was selected as a 10 mg dose (seven days on/off), and dose-dependent hyperphosphatemia was more evident for 4–9 mg QD dosage (50). In the following parts 2–4 of the phase I study (NCT01703481), erdafitinib showed tolerability and clinical activity against FGFR genomic alterations susceptible urothelial carcinoma and cholangiocarcinoma, with the objective response rates of 46.2 and 27.3 %, resp. Hyperphosphatemia, stomatitis, and asthenia of grade 1/2 severity were the most frequently reported side-effects (51). Moreover, no change in QTc interval or other ECG parameters was observed while analyzing the effect of erdafitinib on ECG parameters and the plasma concentration and QTc interval change correlation in advanced or refractory solid tumors (52). In another phase I study on the Japanese population with advanced solid tumors (NCT01962532), erdafitinib was administered on a 2–6 mg QD schedule on a three-week cycle or 10–12 mg intermittently (7 days-on/off) of a four-week cycle (53). Erdafitinib at 10 mg alternate 7 days regimen was well tolerated in Japanese patients and selected as an RP2D for further studies.
Similarly, erdafitinib is being studied in a multicenter, open-label phase II clinical study (NCT02699606) for the treatment of Asian individuals who have advanced non-small cell lung cancer, esophageal cancer, urothelial cancer, and cholangiocarcinoma. Erdafitinib has also been evaluated for safety, pharmacology, and clinical response in Asian patients with hepatocellular carcinoma under phase I/II multicenter investigation (NCT02421185); results are yet to be published. Two phase II clinical studies, NCT03999515 and NCT04754425, sponsored by the University of Washington (Seattle, Washington, USA) and the University of Texas MD Anderson Cancer Center (Houston, Texas, USA), are recruiting patients to evaluate erdafitinib activity in treating castration-resistant prostate carcinoma and double-negative prostate cancer patients, resp. ( Table II). Recently, In an Australian open-label phase II study, erdafitinib enhanced the antitumor effect of androgen deprivation therapy in patients with treatment-naïve prostate cancer (54). However, this combination is not recommended in clinical settings due to patients' poor tolerability.
Table II. Key clinical trials of FDA-approved selective FGFR-TKIs
aInformation assessed from ClinicalTrials,gov; last access date January 23, 2023
Erdafitinib is primarily metabolized by CYP2C9 and CYP3A4 enzymes (55). The participation of CYP2C9 and CYP3A4 enzymes in eliminating erdafitinib is estimated to account for 39 and 20 %, resp., of the total clearance. Erdafitinib, in its original form, remained the primary drug-related component in the plasma, with no detectable metabolites found in circulation. Metabolic clearance was the major elimination mechanism in humans. The primary pathway was O-demethylation to form O-desmethyl erdafitinib metabolite (M6). This metabolite is reported to be a major metabolite in human excreta (24 %) other than unchanged erdafitinib (14.1–22.6 %). Another faecal metabolite is M8, formed via the N-dealkylation pathway. The metabolism scheme of erdafitinib in humans is depicted in Fig. 4. An open-label, parallel-group phase 1 trial (NCT03135106) was conducted to investigate the pharmacokinetics and safety of erdafitinib in healthy volunteers when co-administered with a moderate-to-strong CYP2C9, CYP3A4 and P-glycoprotein inhibitors (fluconazole and itraconazole) (56). Both the inhibitors on co-administration with erdafitinib resulted in increased plasma exposure to erdafitinib in healthy adult participants, potentially necessitating erdafitinib dose reduction, or use of alternative concomitant medicine with little or no CYP2C9 or CYP3A4 inhibition. The concurrent administration of erdafitinib with other medicines that have the potential to influence blood phosphate levels is contraindicated as it may result in an increase or decrease in the levels of serum phosphate. Erdafitinib is a low extraction ratio drug exhibiting high protein binding with alpha-1-acid glycoprotein with varying free fraction concentration across populations (57). Hence, free erdafitinib concentration characterization is critical to account for the difference in free fraction and its clinical relevance across the population.
Pemigatinib
Pemigatinib, also known as INCB054828 and Pemazyre®, is an orally active, potent small molecule, selective and reversible ATP-competitive inhibitor of FGFR1-3 tyrosine kinases discovered by Incyte Corporation (58). Pemigatinib potently inhibits FGFR1-4 kinases with corresponding IC50 values in the 0.4–30 nanomolar range. Pemigatinib is categorized as an FGFR1-3 tyrosine kinase inhibitor that primarily targets FGFR1-3. However, it can also affect FGFR4, albeit only at a much higher dose (30 folds IC50 value) than the other FGFRs. Notably, pemigatinib exhibits a strong affinity for FGFR kinases and does not impact any other protein kinases with IC50 values lower than 1,000 nmol L–1, except for KDR, VEGFR2 and c-KIT, for which the IC50 values are 70, 182, and 266 nmol L–1, resp. In vitro, pemigatinib inhibited Ba/F3 cell lines expressing FGFR1 and FGFR3 kinase domain fusion with IC50 values of 3 and 4 nmol L–1, resp. Preclinically, pemigatinib demonstrated potent antitumor activity in cancer cell lines and tumor xenograft models with FGFR1 (KG1/KG1a, 8p11-positive acute myeloid leukemia), FGFR2 (KATO III, cholangiocarcinoma) and FGFR3 (RT-4/RT112, urothelial carcinoma) translocation with greater sensitivity than cell lines lacking FGFR genetic aberrations (59). Because of its favorable preclinical safety record, pemigatinib was given priority in human clinical trials. The potential inhibition by the drug pemigatinib can be substantiated by an investigation of its molecular interactions and binding orientation within the binding site of FGFR. X-ray solved crystal structure of the FGFR in complex with pemigatinib was extracted from PDB (ID: 7wcl) and analyzed (Fig. 3b) (60). Three characteristic hydrogen bonding interactions were observed between pemigatinib and FGFR. A strong H-bonding (2.9 Å) was noted between the oxygen of methoxy (-OCH3) group on the aromatic ring and an amino group of the acidic amino acid Asp641. Similarly, a couple of other two hydrogen bondings were observed between nitrogens of pyrazolyl and pyridinyl moiety with the crucial aliphatic amino acid Ala564 at a distance of 2.84 Å and 3.0 Å, resp. The heterocyclic core moiety was buried around the residues of Lys482, Leu484, and Tyr563. Moreover, the dimethoxy-substituted phenyl ring showed hydrophobic interactions with amino acids such as Leu630, Val492, and Lys514. All these critical molecular interactions of pemigatinib to FGFR supported the potent inhibition of FGFR in various cancer cell lines.
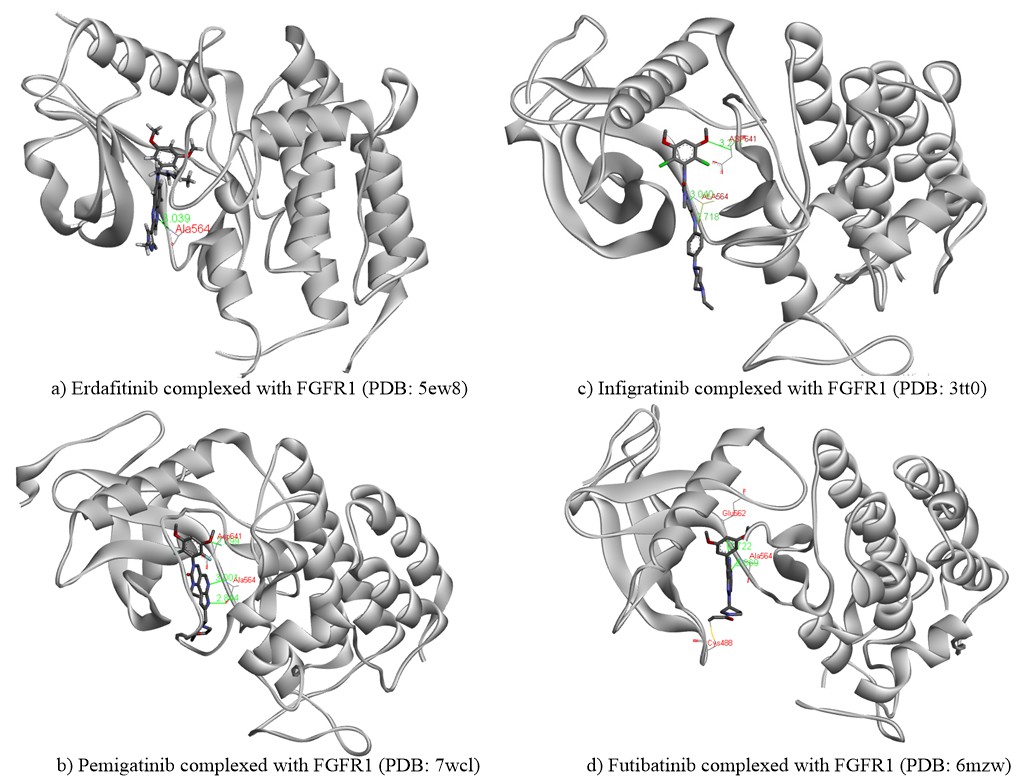
Fig. 3. Binding interactions of a) erdafitinib, b) pemigatinib, c) infigratinib and d) futibatinib to FGFR1. The chemical structures of all four drugs are presented in atom-coloured thick tube model, green coloured lines representing the hydrogen bond, whereas red-coloured text indicates the name of the amino acid involved in crucial hydrogen bonding interaction. The yellow line in d) presents the covalent type of irreversible inhibition between futibatinib and CYS488.
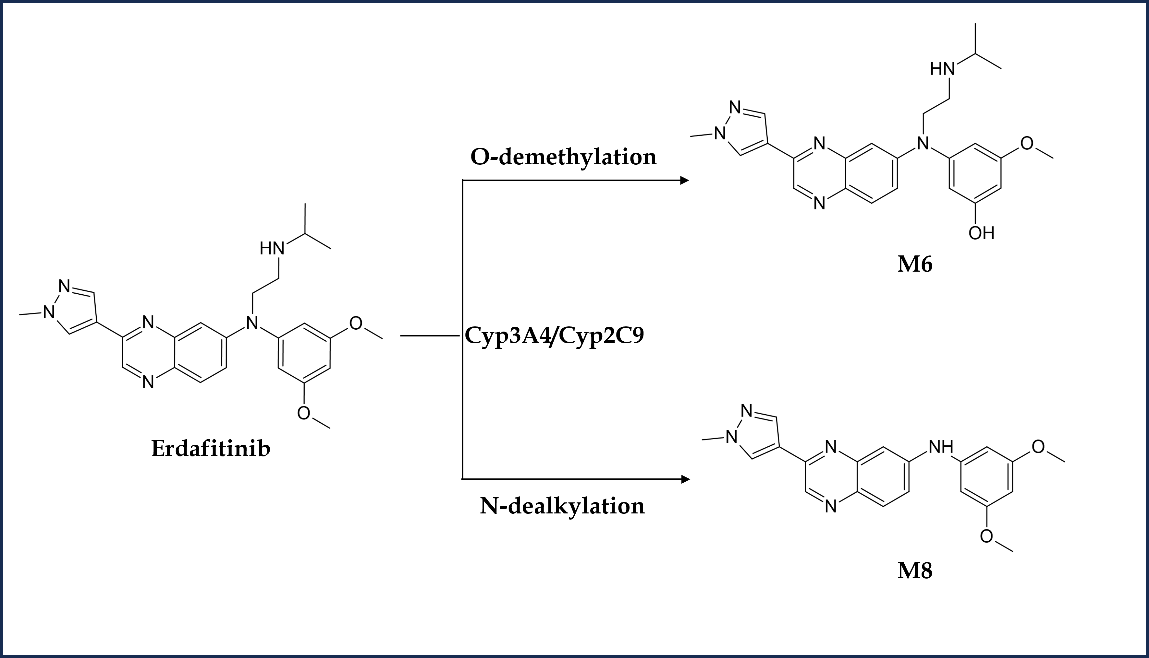
Fig. 4. Metabolism scheme of erdafitinib in humans.
In a two-part pivotal phase I/II study (FIGHT-101, NCT02393248) on patients with refractory advanced malignancies with or without FGFR genetic alterations, pemigatinib demonstrated a double safety profile and pharmacological activity, with higher overall response rate (ORR) in patients with FGFR fusion/rearrangements than in those with FGFR mutations (ORR, n = 5, 25 % vs. n = 3, 23.1 %) (61). Although the MTD was not obtained in the dose escalation part, the pharmacologically active dose was reported to be ≥ 4 mg QD with the terminal t1/2 of 15 h (62). In the part 2 dose expansion study, the RP2D was reportedly 13.5 mg QD, supporting sustained inhibition of FGFR signaling. Alopecia, stomatitis, pneumonia, hyperphosphatemia, tiredness, and dry mouth were the most frequently reported all-cause/grade adverse effects (61). No clinically significant dose-dependent drug effect on heart rate and cardiac repolarization (QTc) was observed in pemigatinib treatment (63).
In another crucial phase II clinical trial (FIGHT-202, NCT02924376) among previously treated 107 patients of locally advanced or metastatic cholangiocarcinoma with or without FGFR2 gene abnormalities, pemigatinib was orally administered at a dose of 13.5 mg QD on a three-week cycle (two weeks on and one week off) until progression of the disease, unacceptable toxicity, physician decision or withdrawal of consent (64). Thirty-eight patients met the primary endpoint, i.e., ORR (36 %), with 3 complete and 35 partial responses. The most frequent adverse effect was hyperphosphatemia; no treatment-related death was reported, irrespective of grade/cause (64). Based on the clinical trial findings, the FDA approved pemigatinib for the treatment of adults with an FGFR2 fusion or other rearrangements harboring metastatic or locally advanced cholangiocarcinoma. FDA has recommended an oral dose of pemigatinib to be 13.5 mg QD for two weeks followed by a one-week interval. For the diagnosis of such FGFR2 fusion or gene rearrangements, the FDA has approved FoundationOne® CDX kit (Foundation Medicine, Inc., Massachusetts, USA.
Table III demonstrates the FDA-approved selective FGFR-TKIs, their target kinases, specific clinical trials, and the approval dates.
In addition, a post hoc analysis of progression-free survival (PFS) found that patients with FGFR2 fusions or rearrangements had a median PFS of 7 months after receiving second-line treatment with pemigatinib in the phase II FIGHT-202 study, despite having previously been treated with first- or second-line systemic therapy for metastatic or advanced cholangiocarcinoma (69). Pemigatinib is also under investigation in a phase III clinical trial (FIGHT-302, NCT03656536) to be evaluated as first-line treatment for patients with FGFR2 rearrangements containing unresectable or metastatic cholangiocarcinoma in comparison with standard chemotherapy (gemcitabine and cisplatin) (70). The FIGHT-302 trial is a global, multicenter study with PFS as the primary endpoint and ORR, response duration, overall survival, safety, and quality of life as secondary endpoints, utilizing an open-label, randomized controlled design. Till the writing of this manuscript, the study is still recruiting patients.
Table III. The FDA-approved selective FGFR-TKI, their target kinases, clinical trials and approval dates
a Information assessed from ClinicalTrials.gov; last access date January 15, 2023
Recently, on August 26, 2022, the FDA approved pemigatinib with a recommended dose of 13.5 mg QD continuously for treating adults with relapsed or refractory myeloproliferative neoplasms (MPN), rare hematologic neoplasms with FGFR1 rearrangements, based on the outcomes of an open-label, multicenter, single-arm, phase II FIGHT 203 clinical trial (NCT03011372) including 28 patients which were either ineligible or relapsed after chemotherapy or allogeneic hematopoietic stem cell transplantation (71).
Pemigatinib treatment continued until unacceptable toxicity, disease progression, or until patients became eligible for allogeneic hematopoietic stem cell transplantation (72). Pemigatinib demonstrated high safety and efficacy in all 28 patients with a complete cytogenetic response rate of 79 % (22/28), suggesting pemigatinib as the first therapy to offer a long-term treatment option for FGFR1 rearrangement MPNs. The most common treatment-related adverse events were hyperphosphatemia, alopecia, stomatitis, diarrhea, anemia, nail toxicity, and ocular toxicity. Pharmacokinetically, pemigatinib is primarily metabolized by CYP3A4, with little renal elimination. Fig. 5 demonstrates that CYP3A4 is solely responsible for the O-demethylation of pemigatinib, forming O-desmethyl pemigatinib (M2). This major metabolite and its secondary metabolites (glucuronide (M7) and sulfate (M9)) account for 76.9 % of the metabolite burden in urine and feces (73). Bis- O-demethylation is another important pathway in the metabolic clearance of pemigatinib in humans. Nevertheless, a dosage adjustment is indicated in patients with severe hepatic or renal impairment, and no dose modification is required in patients with mild hepatic impairment (74). Co-administration of pemigatinib with a strong or moderate CYP3A inducer decreases pemigatinib plasma concentrations, which may reduce the efficacy of pemigatinib. Thus, concomitant use of strong and moderate CYP3A inducers such as itraconazole with pemigatinib is contraindicated (75). Similarly, concomitant use of a moderate CYP3A inducer decreases pemigatinib exposure by more than 50 %. No other clinically significant interactions have been observed with proton pump inhibitors (esomeprazole), histamine-2 antagonists, and OCT2/MATE1 substrates (metformin) on concurrent administration with pemigatinib. Several other phase I/II clinical trials are either completed or are in the active/ recruiting phase for treating cancers with FGFR gene alterations, such as urothelial cancer (NCT02872714, NCT03914794, NCT04294277), pancreas cancer (NCT05216120), colorectal cancer (NCT04096417), non-small cell lung cancer (NCT05004974, NCT05287386, NCT05253807), breast cancer (NCT05560334), gastrointestinal cancer (NCT05559775, NCT05202236, NCT05529667), glioblastoma (NCT05267106), endometrial cancer (NCT04463771), and acute myeloid leukemia (NCT04659616) (Table II).
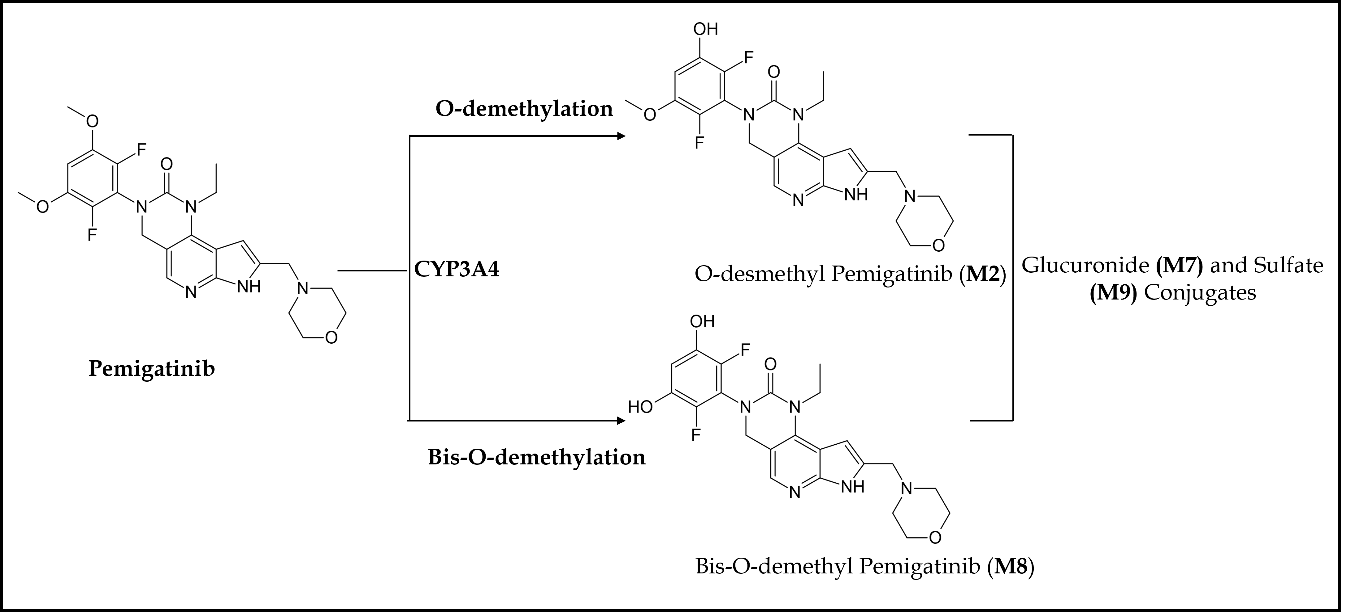
Fig. 5. Main metabolic pathways of pemigatinib in humans.
Infigratinib
Infigratinib, also referred to as BGJ398 and TRUSELTIQTM, is an orally active, ATP-competitive, selective FGFR1-4 kinase inhibitor having IC50 values in the range of 1.1–61 nmol L–1 (66). IC50 values for other kinases were >1000 nmol L–1 except for VEGFR2, KIT and LYN. Infigratinib was initially developed by QED Therapeutics, Palo Alto, California, USA, as an anticancer agent against bladder cancer with FGFR kinase genetic alterations. In vivo, this pharmaceutical agent demonstrated significant antitumor activity against wild-type FGFR3 overexpressing RT112 bladder cancer xenograft models (76). Subsequently, high-throughput cell line profiling revealed that infigratinib inhibits FGFR signaling and reduces cell growth in cancer cell lines with FGFR genetic mutations (77). An examination of the complex of infigratinib in the FGFR binding site at a molecular level (PDB ID: 3TT0) revealed significant molecular interactions responsible for its anticancer potential (76). According to Fig. 3c, the phenylamine NH and the pyrimidine ring nitrogen in infigratinib demonstrated crucial hydrogen bonding interactions with the carbonyl oxygen and the amino groups of the significant amino acid Ala564 at a distance of 2.72 and 3.04 Å, resp. Similarly, one more hydrogen bonding (3.21 Å) was observed between the oxygen of the methoxy group on the aromatic ring and the amino group of the acidic amino acid Asp641. Hence, these three hydrogen bonding interactions contributed majorly towards the anticancer activity of infigratinib through inhibition of FGFR. Further, the dimethoxy-substituted aromatic ring was placed within the pocket surrounded by amino acids such as Leu484, Val492, Glu562, and Tyr563 of the binding site, thus conferring the selectivity of the drug against FGFR.
Clinically, in a global first-in-human phase I study in the patients with advanced solid tumors possessing FGFR gene alterations (amplifications, mutations, or fusions), infigratinib treatment was tolerable, and the maximum tolerated dose (MTD)/RP2D was determined to be 125 mg QD (3-weeks-on and 1-week-off cycle) (78). Moreover, infigratinib demonstrated antitumor activity in FGFR alterations driven squamous non-small cell lung cancer (FGFR1 amplification) , cholangiocarcinoma (FGFR2 fusion/mutation), and bladder cancer/urothelial carcinoma (FGFR3 mutations), supporting parent clinical investigations in phase II study.
Similarly, a phase Ib study (NCT02257541) focused on investigating the safety of infigratinib with imatinib mesylate in advanced gastrointestinal stromal tumor (GIST); toxicity was encountered with the combination therapy leading to early withdrawal of the study before identification of RP2D or dosing schedule (79).
In a multicentric, open-label phase II clinical trial (NCT02150967) on chemotherapy-refractory 108 patients with unresectable, locally advanced, or metastatic cholangiocarcinoma containing FGFR2 fusion or rearrangements, infigratinib was administered 125 mg QD for continuous three weeks followed by one week off (four-week cycle), until disease progression or worsening of the symptoms (80). The pharmaceutical agent showed a BICR-assessed ORR of 23 % (25/108) with one complete response and 24 partial responses. No death was reported; the most common adverse events were hyperphosphatemia, stomatitis, fatigue, alopecia, and dry eyes (81). Based on these encouraging findings, the FDA on May 28, 2021, granted infigratinib accelerated approval for treating previously treated patients with unresectable, advanced, or metastatic cholangiocarcinoma with an FGFR2 fusion or rearrangements, identified explicitly by the FDA-approved FoundationOne® CDX test (Foundation Medicine, Inc., Massachusetts, USA) (66). The recommended dose of infigratinib is 125 mg QD for 21 days continuously, followed by 7 days off (28 days cycle).
Likewise, a multisite, randomized, open-label phase III PROOF 301 (NCT03773302) clinical study is underway to investigate infigratinib in the front-line setting compared to standard-of-care chemotherapy (cisplatin and gemcitabine) in advanced or metastatic cholangiocarcinoma containing FGFR2 translocations (82). The efficacy and antitumor activity of orally administered infigratinib (125 mg QD for 3 weeks every 28 days) was also appraised on previously treated patients ( n = 67) with advanced urothelial cancer containing FGFR3 alterations in phase II prospective, open-label, clinical trial. Infigratinib achieved its primary endpoint with an ORR of 33.3 % and a disease control rate of 75 %. The most common undesirable effect was hyperphosphatemia (78 %, 48/67 patients) (83). The safety and effectiveness of infigratinib as adjuvant therapy for patients with FGFR3 genetic alterations driven invasive urothelial carcinoma are now being inspected in a multipoint, randomized, double-blind, placebo-controlled, phase III clinical trial (PROOF-302, NCT04197986); the primary outcome measure is centrally determined disease-free survival (84).
Similarly, a multicenter, single-arm, open-label, phase II trial evaluated the effectiveness of infigratinib in FGFR-altered recurrent gliomas (85). When used as monotherapy, infigratinib produced limited efficacy in patients but had a stable disease control lasting more than a year. Hyperphosphatemia was the most common treatment-related side-effect. Several other indications are being investigated for infigratinib in multiple phase I/II clinical trials, such as achondroplasia (NCT04035811, NCT04265651, NCT05145010), gastric cancer, and gastroesophageal junction adenocarcinoma (NCT05019794) and BRAF-melanoma (NCT02159066) ( Table II.
Pharmacokinetically, infigratinib is primarily metabolized by CYP3A4. Based on in vitro research, it has been observed that around 94 % of infigratinib undergoes metabolism mediated by the enzyme CYP3A4, while approximately 6 % of the drug is metabolized by flavin-containing monooxygenase 3 (FMO3) (66). Approximately 38 % of the administered dose of infigratinib is present as unchanged in the plasma. Additionally, two significant metabolites of infigratinib, viz., BHS697 and CQM157, are detected at levels exceeding 10 % of the administered dose. BHS697 and CQM157 exhibit pharmacological action in addition to infigratinib, with BHS697 accounting for around 16 to 33 % of the overall pharmacological activity, while CQM157 contributes approximately 9 to 12 %. BHS697 is subject to additional metabolic processes facilitated by the enzyme CYP3A4, while CQM157 undergoes biotransformation via both phase I and phase II routes. Further investigation is required to properly characterize the precise metabolic pathways and structural properties of BHS697 and CQM157. Recently, an in vitro and in vivo metabolic profiling study in rats elucidated the involvement of various phase I and II bioactivation pathways for infigratinib metabolism (86). Phase I pathways include hydroxylation, N-demethylation, dechlorination, O-demethylation, and N-dealkylation (Fig. 6). Sulfation and glucuronic acid conjugation were the main phase II pathways for infigratinib metabolism. N-ethyl piperazine ring and 2,6-dichloro-3,5-dimethoxyphenyl ring of the infigratinib were reported to be the key sites of metabolism. In vitro bioactivation assessment also revealed the formation of two highly reactive intermediates, such as iminium ions and 1,4-benzoquinones, suggesting a possible mechanism of infigratinib toxic effects. Individuals with abnormal renal or hepatic function had a higher corresponding potency adjusted steady-state AUC for infigratinib and its metabolic products; hence, dose reduction is indicated in patients with mild-to-moderate renal or hepatic impairment to avert unwanted toxicity (66). CYP3A primarily metabolizes infigratinib. Hence, concurrent use of infigratinib with strong or moderate inducers or inhibitors of CYP3A may decrease or increase the plasma concentration of infigratinib, compromising its antitumor activity. Similarly, infigratinib should not be administered with gastric acid-reducing agents such as proton pump inhibitors and histamine-2 antagonists due to their plasma concentration decreasing potential.
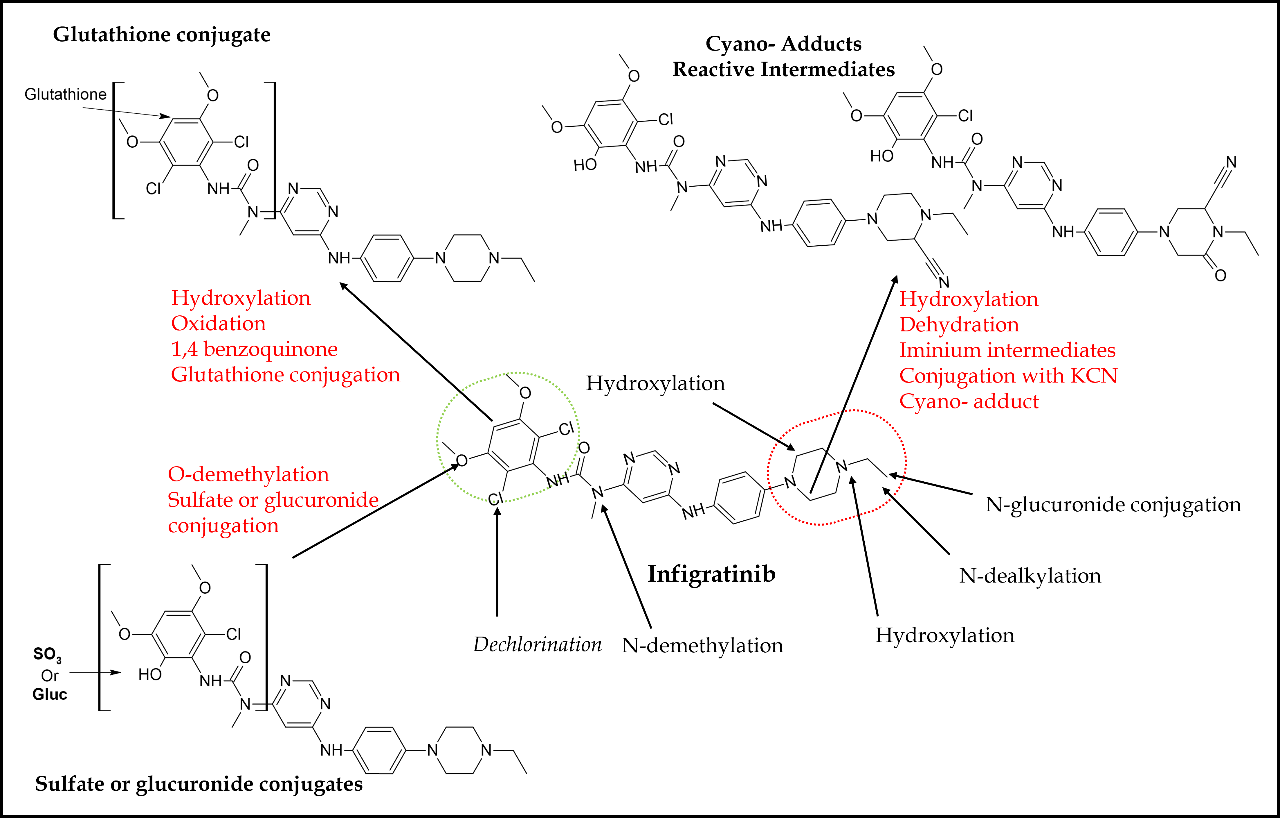
Fig. 6. In vitro and in vivo bioactivation pathways and reactive metabolites of infigratinib in rats.
Futibatinib
Futibatinib also recognized as TAS-120 and LYTGOBI®, is an oral, potent, selective, covalently bound, and irreversible small molecule inhibitor of FGFR1-4 kinase with IC50 values ranging from 1.8 to 3.7 nmol L–1 (87, 88). Taiho Oncology, Princeton, New Jersey, USA, and Taiho Pharmaceutical, Tokyo, Japan discovered futibatinib as a potential therapy for individuals suffering from advanced tumors carrying FGFR1-4 genetic anomalies, including cholangiocarcinoma, who had previously been treated with chemotherapeutic agents or other anticancer treatments (67). Futibatinib preferentially and irreversibly links to the ATP binding region of FGFR1-4, disrupting FGFR-mediated pathways of signal transduction, decreasing tumor proliferation, and promoting tumor cell death in cancers with FGFR1-4 genetic variations. A molecular binding interaction analysis of futibatinib with FGFR was investigated using the X-ray-resolved co-crystallized complex acquired from PDB (ID: 6MZW) (88). According to Fig. 3d, the free primary amino group on the pyrimidine ring showed a strong hydrogen bonding interaction with the oxygen atom of the acidic amino acid Glu562 at a distance of 2.72 Å, whereas the heterocyclic nitrogen of pyrimidine interacted with the crucial amino acid Ala564 by forming a significant hydrogen bond at a distance of 2.87 Å. Both of these interactions are observed in the hinge region of the active-site amino acids of FGFR. Further, the dimethoxy-substituted aromatic ring was buried within the pocket surrounded by hydrophobic aliphatic amino acids such as Val492, Lys514, Val559, and Leu630. On the other side, the pyrrolidine side chain of futibatinib was enclosed by the residues of Leu484, Gly485, Val492 and Tyr563. The major reason for the irreversible covalent inhibition of FGFR by futibatinib is the presence of acrylamide moiety and in particular, the terminal carbon of acrylamido moiety in futibatinib exhibited a characteristic covalent interaction with the sulfur atom of the amino acid Cys488. This could be possible only if the pyrrolidine ring is positioned upward direction to meet P-loop active site amino acids. It is also very important to note that Cys488 is an amino acid from P-loop amino acids and its interaction with futibatinib has been confirmed through experimental mass spectrometry (LC-MS/MS fragmentation) analysis. This has been further supported by an investigation into the extreme kinetics of the reactivity by X-ray crystallography experiments. It was deduced that the acrylamide moiety of futibatinib confers exclusively a cis configuration while interacting with FGFR by forming an irreversible drug-receptor complex. Thus, the “bent” P-loop approaches closely to the acrylamide moiety of futibatinib to tolerate the thia-Michael addition type of reaction. The scheme with relevant chemical reaction is presented in Fig. 7. Preclinically, futibatinib showed potent and selective growth inhibition ( GI50 ranging from 1 to ∼50 nmol L–1) of a wide range of malignant cell lines inhibiting FGFR genetic aberrations, such as gastric, myeloma, lung, endometrial, bladder and breast cancer cell lines by impeding phosphorylation of FGFR and MAPKs and PI3K/Akt downstream signaling pathways. Similarly, in vivo futibatinib in oral administration resulted in dose-dependent tumor shrinkage in carcinogenic FGFR-driven human xenograft carcinoma models, supporting clinical exploration in individuals with FGFR-driven cancers (87).
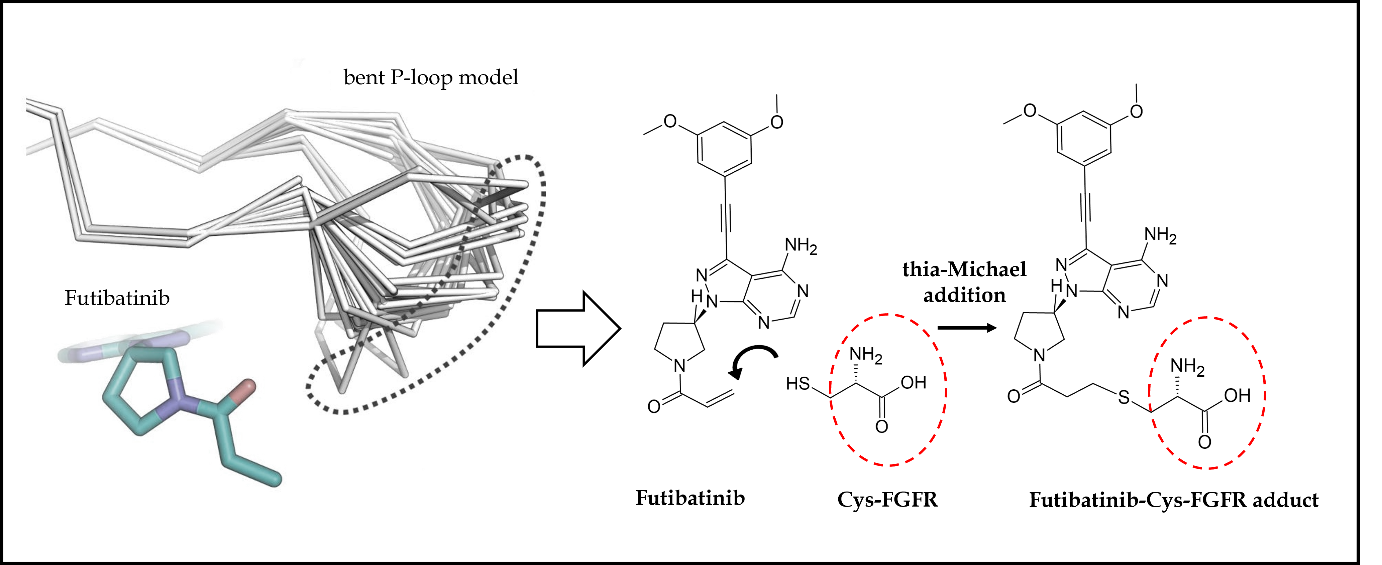
Fig. 7. Futibatinib-FGFR1 covalent interaction: cysteine (Cys488) amino acid in the bent P-loop conformation exhibits movement (dot-covered region) to suitably interact with acrylamide moiety of futibatinib through Michael addition reaction mechanism, thus, resulting in strong irreversible covalent bonding interaction.
In a phase I dose escalation study (FOENIX-101, NCT02052778), futibatinib's safety, efficacy, and pharmacokinetic/pharmacodynamics were evaluated in individuals ( n = 86) with advanced solid tumors expressing FGF/FGFR aberrations (89). Futibatinib administration displayed satisfactory safety and pharmacodynamic effects in adults with MTD and RP2D at 20 mg QD; however, the 24 mg QD dose indicated dose-limiting toxicity. The most common adverse reactions documented were hyperphosphatemia, constipation, and diarrhea). Similarly, in a phase I dose-expansion study on 197 participants with an advanced solid tumor, futibatinib demonstrated broad-spectrum antitumor activity in an array of tumors (gastric, cholangiocarcinoma, urothelial, head and neck, urothelial and breast cancer) harboring both recognized and uncharacterized FGFR1-3 genetic alterations (90). The ORR was 13.7 %, with the highest activity against FGFR2 fusion or rearrangement-bearing cholangiocarcinoma (ORR, 25.4 %).
Based on favorable results from the phase I study, futibatinib advanced to a decisive phase II clinical trial (FOENIX-CCA2) in which participants with unresectable, metastatic cholangiocarcinoma bearing FGFR2 fusion/rearrangements and had disease progression even after more than one prior chemotherapy session were administered with futibatinib (20 mg QD) until progression of the disease or intolerability (91). Across 103 patients, the ORR was 42 % (43/107), with all examinees achieving partial responses. Responses were durable; the median duration of response was 9.7 months. Commonly documented treatment-related adverse events were hyperphosphatemia, alopecia, dry mouth, stomatitis, nail toxicity, dysgeusia, dry eye, constipation, diarrhea, musculoskeletal pain, abdominal pain, dry skin, urinary tract infection, fatigue, decreased appetite, arthralgia, nausea, and vomiting. Subsequently, on September 30, 2022, the FDA approved futibatinib, with a recommended dose of 20 mg QD for treatment-experienced, unresectable, advanced/metastatic cholangiocarcinoma patients bearing various FGFR2 genetic abnormalities, including rearrangements and fusions (67).
In vitro pharmacokinetics studies on futibatinib revealed that it is metabolized by CYP3A4 like other FGFR TKIs (92). Primary metabolic pathways of futibatinib were evaluated in a phase I study using14C-futibatinib single oral 20-mg dose in healthy participants. Unchanged futibatinib was reported to be a major circulating constituent in the plasma, accounting for almost 60 percent of circulating radioactivity. However, a negligible concentration of parent futibatinib was observed in feces and urine, indicating complete metabolism before elimination. This agent is primarily metabolized by cytochrome P450 enzyme-mediated O-demethylation as the main oxidation pathway and glutathione conjugation (Fig. 8). Cysteinylglycine-conjugated futibatinib and desmethyl futibatinib were the most abundant metabolites in plasma and feces, resp. Similarly, glucuronide and sulfate of desmethyl futibatinib and glutathione- and cysteine-conjugated futibatinib were the key metabolites in human hepatocytes. It takes a median time of 2 hours to reach maximum plasma concentration ( tmax), and AUC and cmax were observed to be reduced by 11 and 42 %, resp., in healthy individuals after consuming a high-fat, high-calorie meal. However, futibatinib may be administered with or without food. Almost 91 % of the radioactivity excreted after a single oral dose of radioisotope-tagged futibatinib (20 mg) was found in the stool, while only nine percent was found in the urine, and there was almost no unchanged futibatinib in either form (67, 88, 93). Owing to the CYP3A and P-gp inhibitory potential of futibatinib, the drug-drug interactions of futibatinib with itraconazole (a dual P-gp and strong CYP3A inhibitor), rifampin (a dual P-gp and strong CYP3A inducer), or midazolam (a sensitive CYP3A substrate) in healthy adult participants were investigated in a phase I study. The study results suggest that coadministration of dual P-gp and strong CYP3A inhibitors or inducers with futibatinib should be avoided (94). However, futibatinib can be concomitantly administered with other drugs metabolized by CYP3A. In another drug-drug and drug-food interaction investigation, no significant differences in futibatinib exposure between futibatinib plus lansoprazole and futibatinib alone were observed (95). Similar interactions were observed with food, suggesting futibatinib may be used with or without food and concomitantly with proton pump inhibitors.
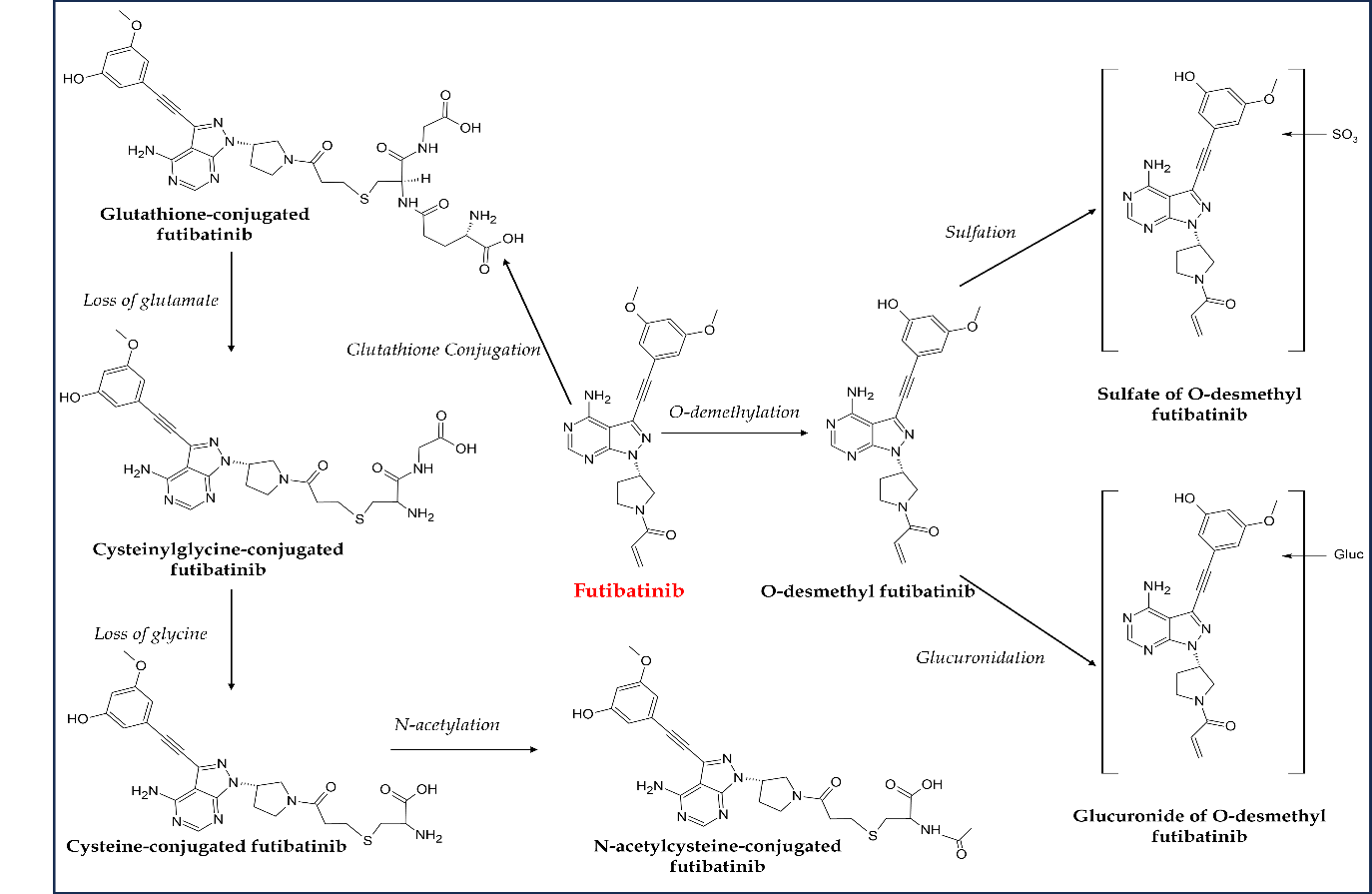
Fig. 8. Major metabolic pathways of futibatinib in humans.
Several other clinical studies investigate futibatinib for treating various FGFR genomic aberrant-driven tumors. In addition to the currently underway phase II clinical study in cholangiocarcinoma (based on which FDA approval was granted), an open-label, global, parallel two-arm, randomized phase III clinical trial (FOENIX-CCA3, NCT04093362) is recruiting patients with advanced cholangiocarcinoma bearing FGFR2 gene rearrangements. The primary outcome measure is PFS. Another phase III trial in patients with advanced biliary cancer is recruiting patients with a primary endpoint and secondary endpoint PFS and ORR, resp. Multiple phase I and II studies are also at the recruitment stage for investigating futibatinib safety and efficacy either as monotherapy or in combination with chemotherapeutics in FGFR-driven cancers like advanced KRAS mutant cancer (NCT04965818), advanced or metastatic urothelial cancer (NCT04601857), advanced hepatocellular carcinoma (NCT04828486), metastatic microsatellite stable endometrial carcinoma (NCT05036681) and metastatic breast cancer (NCT04024436) ( Table II).
FGFR INHIBITORS AS ANTICANCER MODALITY
Cholangiocarcinomas are rare, genetically heterogeneous, and highly aggressive cancers with incidence rates below 6/100,000. FGFR2 fusions are an emerging class of drug targets in cholangiocarcinoma oncology, detected in almost 10–15 % of patients (96). Due to the late onset of clinical symptoms, the majority of patients arrive with locally advanced or metastatic conditions, and most have rapid recurrence even after complete resection. Therefore, palliative treatments with chemotherapeutics are the mainstay of cholangiocarcinoma therapy (97). However, the first-line treatment with chemotherapy offers limited benefits due to higher incidences of chemotherapy-refractory cholangiocarcinoma with a median overall survival rate of 11–13 months. Efficient second-line treatments are lacking. Based on promising results reported from various phase II clinical studies, FGFR2 fusions, one of the common generic alterations associated with cholangiocarcinoma, have become promising targets for precision therapeutics, highlighting the importance of FGFR2-directed therapy as a meaningful second-line treatment of cholangiocarcinoma (98). The FDA has approved two reversible, selective FGFR inhibitors (pemigatinib, infigratinib) and one irreversible selective inhibitor (futibatinib) for the locally advanced and metastatic cholangiocarcinoma as the second-line treatment. All three approvals came from the results of phase II clinical trials. Further clinical trials are comparing the effect of these approved FGFR-TKI with chemotherapeutics, like cisplatin plus gemcitabine, as a potential first-line or rescue treatment (NCT03656536, NCT03773302, NCT04093362).
Several other small molecule FGFR inhibitors, including erdafitinib, derazantinib, and Debio 1347, are under investigation for their efficacy in cholangiocarcinoma malignancies (99). Derazantinib, a multi-kinase inhibitor with FGFR1-4 inhibitory potential, in phase I/II clinical investigation (NCT01752920), has displayed good safety and efficacy in chemo-refractory, unresectable, and advanced cholangiocarcinoma patients with FGFR2 fusion. Another pivotal single-arm phase II trial (NCT03230318) on 148 patients with inoperable or advanced cholangiocarcinoma with FGFR2 genetic aberrations has been completed, but results are yet to be published.
Urothelial carcinoma, the sixth most common cancer in the USA, is known to encompass divergent molecular alterations and morphologic subtypes (100). The FGFR pathway is a recognized oncogenic driver in urothelial cancer, leading to a new era of targeted drug development. FGFR3 gene alterations are common in urothelial cancer, with a higher rate of incidences in the upper tract than in lower tract cancers. As a result, several FGFR-directed targeted treatments have been developed and tested in clinical trials with urothelial cancers and other solid tumors with FGFR mutations (101). Erdafitinib is one such FDA-approved FGFR inhibitor for the treatment of platinum chemotherapy-refractory metastatic urothelial cancer containing FGFR2/3 genetic alterations. Other FGFR-directed inhibitors, including derazantinib (NCT04045613), ASP5878 (NCT02038673), PRN1371 (NCT02608125), rogaratinib (NCT03410693) and AZD4547 (NCT05086666) are under investigation in various phase I or II clinical studies, either alone or in combination with immunotherapeutics for oncogenic FGFR-driven urothelial carcinoma. Among these inhibitors, rogaratinib, a pan-FGFR inhibitor, showed comparable safety and efficacy in an open-label, randomized phase II/III clinical trial (FORT-1, NCT03410693) on comparing chemotherapy with FGFR-directed therapy in FGFR1/3 mRNA overexpression positive urothelial carcinoma patients (102).
Myeloid/lymphoid neoplasms with FGFR1 rearrangements are rare, hematologically and genetically heterogeneous malignancies with eosinophilia as the most frequent presentation (103). Recently, pemigatinib became the first FDA-approved targeted treatment for patients with FGFR1 gene arrangements in relapsed or refractory myeloid/lymphoid neoplasms.
PAN-FGFR/SELECTIVE FGFR INHIBITORS
In addition to the reviewed selective FGFR inhibitors, several other FGFR-TKIs are under development and investigated in different phases of clinical trials, including selective FGFR inhibitors (pan-FGFR inhibitors, FGFR1-3 inhibitors, and FGFR4 inhibitors), covalent FGFR inhibitors, and non-selective multi-kinase inhibitors (104). Furthermore, some of them are showing promising results in clinical trials. Surprisingly, all four approved FGFR inhibitors are pan-FGFR inhibitors. Other pan-FGFR inhibitors under investigation in different phases of clinical trials include PRN1371 (NCT02608125), ASP5878 (NCT02038673), LY2874455 (NCT01212107), AZD-4547 (NCT04439240) and rogaratinib (NCT03410693). Similarly, various FGFR1,2,3 inhibitors are also under investigation, for example, Debio1347 (NCT03344536), E7090 (NCT04238715), and HMPL-453 (NCT05173142). Debio 1347 showed promising efficacy and tolerability up to 80 mg per day in a phase I study with advanced solid tumor patients with FGFR genetic alterations (105). Recently a multicenter, investigator-initiated phase II clinical trial (NCT04962867) was designed and registered to assess the safety and efficacy of E7090 (tasurgratinib) in advanced or recurrent solid tumors containing FGFR genetic alterations (106).
A few non-selective multi-kinase inhibitors are directed against FGFR-driven malignancies. Dovitinib, a pan-kinase inhibitor of VEGFR, FGFR, PDGFR-beta, and c-KIT, was investigated in a phase II non-randomized trial (NCT01379534), as second-line therapy in advanced or metastatic endometrial cancer patients with FGFR2 mutation; in the results, it did not meet the prespecified endpoints (107). However, another multi-kinase inhibitor, derazantinib, could potentially treat cholangiocarcinoma with a mutated FGFR2 gene in separate phase I/II clinical trials (NCT01752920) (99).
Hepatocellular carcinoma is reported to harbor aberrant FGF19-FGFR4 signaling in almost 10–20 % of cases (108). Moreover, amplified FGF19-FGFR4 signaling is linked with disease progression and unfavorable prognosis in clinic-pathological examinations, suggesting FGFR4 is the suitable target for hepatocellular carcinoma treatment. Owing to frequent overexpression of FGFR4 in the pathogenesis of hepatocellular carcinoma, several selective FGFR4 inhibitors have been developed and pushed into different phases of clinical trials, including ISIS-FGFR4RX (NCT02476019), fisogatinib (BLU-554, NCT02508467, NCT04194801), roblitinib (FGF-401, NCT02325739), U3-1784 (NCT02690350) and H3B-6527 (NCT02834780) (109). Among these inhibitors, ISIS-FGFR4RX and fisogatinib have entered the phase II stage of clinical trials for obesity and hepatocellular carcinoma, resp. Recently, in a first-in-human phase I trial, roblitinib showed a promising safety profile in FGFR4/KLB+ hepatocellular carcinoma or advanced solid tumors patients, both monotherapy and combined with spartalizumab, an anti-PD-1 antibody (110). However, none of these FGFR4 inhibitors has been approved for clinical use. Future research in advanced clinical trial phases with these inhibitors can revolutionize the therapeutic options for FGFR4 gene-altered carcinomas, including hepatocellular cancer.
RESISTANCE AND ADVERSE EVENTS TO FGFR INHIBITORS
According to many preclinical and clinical research findings, resistance to FGFR TKIs has emerged as a significant challenge. Resistance has been attributed to various mechanisms: gatekeeper mutation-induced steric clashes, alternative signaling pathways feedback activation (RAS-MAPK, PI3K-AKT), lysosome sequestration-mediated TKI sequestration, and gene fusions (104). Moreover, several studies have shown acquired resistance in patients taking non-covalent FGFR inhibitors such as erdafitinib and infigratinib on disease progression. Futibatinib is the only approved covalent-irreversible inhibitor reported to overcome FGFR2 mutations related to Debio 1347 or infigratinib-associated acquired resistance in patients with FGFR2 gene-altered cholangiocarcinoma (111). Thus, covalent or extracellularly acting inhibitors, or even combination treatment with other onco-therapeutic agents, may be worth investigating for their potential to reduce FGFR-TKI resistance.
Across all four FDA-approved FGFR inhibitors, the most common adverse events reported were hyperphosphatemia, alopecia, stomatitis, nail changes, ocular disorders, skin changes, dry mouth, diarrhea, asthenia, fatigue, and nausea, irrespective of disease type. Although, as per clinical results, all the adverse effects are manageable, certain cases may require dose adjustment or discontinuation of therapy. Hyperphosphatemia is a well-understood, mechanism-based toxicity of FGFR inhibitors and is suggested to be a predictive biomarker for FGFR-TKI treatment response. It is usually reversible and easily manageable. In summary, a thorough understanding of unique side effects and proper sensitization of patients about side effects may help prevent unnecessary treatment interruptions and dose adjustments.
CONCLUSIONS
Deregulated FGF/FGFR signaling is regarded as a driver in multiple advanced carcinomas for their proliferation, invasion, and migration mediated by various FGF/FGFR genomic aberrations. Comprehensive genomic profiling has shown frequent tumor FGFR alterations in multiple cancers, including genetic amplifications, mutations, rearrangements, and fusions. FGFR targeting has been established as a potential therapeutical target leading to several FGFR inhibitors, either in therapy or in the developmental pipeline to address these altered receptors as a novel drug category.
The present review discussed four FGFR inhibitors that received FDA approval for malignancies, including cholangiocarcinoma, urothelial carcinoma, and myeloid/lymphoid neoplasms. These agents are erdafitinib, pemigatinib, infigratinib, and futibatinib. The kinase inhibitory activity and pharmacological applications of four drugs were also summarized. In addition, all four drugs showed CYP3A4-mediated metabolism in in vitro pharmacokinetic studies. Pemigatinib is the only FGFR inhibitor that has also been approved by EMA and PMDA on March 26, 2021, and March 23, 2021, resp., for treating FGFR2 fusion or other rearrangements containing locally advanced or metastatic cholangiocarcinoma. Moreover, pemigatinib is the only drug approved for multiple therapeutic indications, viz., cholangiocarcinoma and myeloid/lymphoid neoplasms. Further investigation in this area may open new horizons in genetically altered cancer treatment.
Abbreviations, acronyms, symbols. – ABL1 – tyrosine-protein kinase ABL1, AKT – protein kinase B, ATP – adenosine triphosphate, BICR – blinded independent central review, BLK – B-cell lymphocyte kinase, c-KIT – receptor tyrosine kinase type III, CYP – cytochrome P450, ERK – extracellular signal‑regulated kinase, FGFR – fibroblast growth factor receptor-tyrosine kinase, FLT-3 – FMS-like tyrosine kinase 3, GAB1 – GRB2 associated binding protein 1, GRB2 – growth factor receptor-bound protein 2, JAK – Janus kinase, KDR – kinase insert domain receptor, KG1/KG1a – acute myelogenous leukemia cell lines, LCK – lymphocyte-specific protein tyrosine kinase, LYN – LCK/Yes, novel tyrosine kinase, MAPK – mitogen-activated protein kinase, MEK – mitogen-activated protein kinase kinase, mTOR – mammalian target of rapamycin, PDGF – platelet-derived growth factor, PDGFRA – platelet-derived growth factor receptor alpha, PDGFRB – platelet-derived growth factor receptor beta, PI3K – phosphoinositide 3-kinase, PKC – protein kinase C, PLC-γ – phospholipase C gamma 1, QD – once-a-day, RAF – rapidly accelerated fibrosarcoma, RAS – rat sarcoma virus, RET – rearranged during transfection tyrosine kinase receptor, SOS – son of sevenless protein, STAT – signal transducer and activator of transcription, TIE1 – tyrosine kinase with immunoglobulin-like and EGF-like domains 1, VEGF – vascular endothelial growth factor, VEGFR – vascular endothelial growth factor receptors, VEGFR2 – vascular endothelial growth factor receptor 2.
Acknowledgements. – Hafiz A. Makeen extends his appreciation to the Deputyship for Research & Innovation, Ministry of Education in Saudi Arabia for funding this research work through project number ISP22-2.
Conflict of interest. – The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Authors contributions. – Conceptualiation and research design, N.K-J., N.T. and H.A-M.; writing original draft preparation, N.K-J., M.T., N.T., M.A-B., A.N., M.T., K.Z., H.K-J. and H.A-A.; review and editing, H.A-M., M.A-B., A.N., H.A-A., M.A., B.C. and K.Z. All authors have read and agreed to the published version of the manuscript.