
Introduction
Changing and often extreme environmental conditions are the main cause of abiotic stress and pose a major challenge to plant survival. The inability of plants to relocate exacerbates this challenge. Abiotic stress has negative effects on essential plant processes such as metabolism, growth, and development, as has been extensively documented (Lichtenthaler 1998, Shulaev et al. 2008, Zandalinas et al. 2022). Primary sources of climate-related abiotic stress such as drought, salinity, heat stress, cold, and flooding lead to an overproduction of reactive oxygen species (ROS) in plant cells. ROS include molecules such as hydrogen peroxide (H2O2), superoxide ions (O2•–) and hydroxyl radicals (OH•), which act as signalling molecules in triggering the cellular response to stress (Mandal et al. 2022). At high concentrations, however, ROS outweigh the enzymatic and non-enzymatic antioxidant system and put the cell in a state of oxidative stress (Cramer et al. 2011, Mandal et al. 2022), which leads to the inactivation of cellular mechanisms and, ultimately, to the death of the plant cell.
Epigenetic mechanisms enable plants to respond quickly and precisely to changes in abiotic conditions in the environment. When plants are exposed to different sources of abiotic stress, changes in methylation patterns and chromatin modifications occur both at individual loci and globally at the whole genome level (Zhang et al. 2018, Liu and He 2020); in some cases, meiotic inheritance of stress-induced changes is transmitted to subsequent generations (Liu and He 2020, Ramakrishnan et al. 2022).
This review elucidates the basic epigenetic mechanisms utilised by plants exposed to abiotic stress, thus enhancing our understanding of these intricate processes. By altering DNA methylation patterns and histone modifications, plants can dynamically regulate gene expression in response to different environmental conditions. Small non-coding RNAs, such as small interfering RNAs (siRNAs), are also involved in the fine-tuning of gene expression and mediate RNA-directed DNA methylation (RdDM) to silence transposable elements (TEs) under stress conditions. A comprehensive understanding of these mechanisms and their associated functions (Fig. 1) holds considerable potential for further development of epigenetic-based plant protection strategies and the production of crops that are resistant to abiotic stress.
Epigenetic code in plants
Epigenetic changes include all changes in the chromatin structure that lead to increased or decreased gene expression without altering the underlying DNA sequence. The complex combination of different epigenetic modifications and factors that collectively regulate gene expression and cellular identity, known as the epigenetic code, consists of histone variants and histone modifications (methylation, acetylation, phosphorylation, ubiquitination and SUMOylation), DNA methylation and non-coding RNAs (Duan et al. 2018, Chung et al. 2022). The effects of epigenetic control on gene activity depend on the specific types of epigenetic modifications and their proximity to neighbouring genes (Chang et al. 2020). The presence of heterochromatin marks such as DNA methylation and the methylated histone variant H3K9me2 can have an inhibitory effect on the expression of downstream genes if the marks are positioned within the promoter (Lei et al. 2015, Shi et al. 2023). Furthermore, in some cases, these features within the gene body can impair the efficiency of full-length transcript generation (Wang et al. 2013a, Duan et al. 2017). In addition, epigenetic marks of DNA and histone on TEs located within the promoters of stress-responsive genes play an important role in the dynamics of stress response regulation. By activating TEs under stressful conditions, these marks lead to upregulation of stress-related genes and facilitate adaptive responses to environmental challenges (Chang et al. 2020).
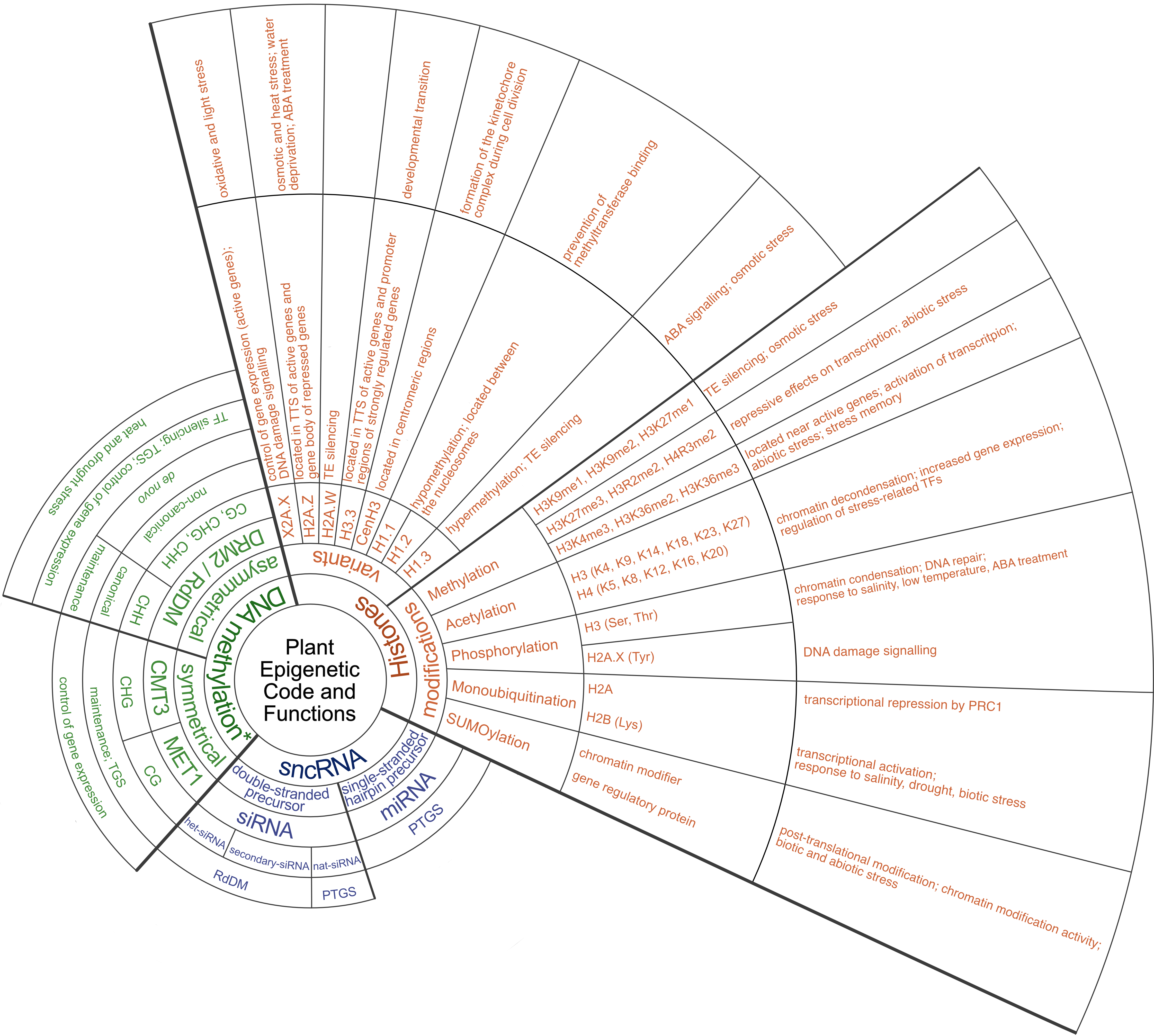
Fig. 1. Schematic diagram illustrating epigenetic mechanisms involved in plant stress response. The diagram includes histone variants and modifications, DNA methylation, and small non-coding RNAs, each annotated with their putative functions in stress response. Abbreviations: ABA – abscisic acid; CMT3 – Chromomethylase 3; DRM2 – Domains rearranged methyltransferase 2; Lys – Lysine; MET1 – Methyltransferase 1; miRNA – micro RNA; PRC1 – Polycomb repressive complex 1; PTGS – Post-transcriptional gene silencing; RdDM – RNA-directed DNA methylation; ROS1 – Repressor of Silencing 1; Ser – Serine; siRNA – small interfering RNA; sncRNA – small non-coding RNA; TE – Transposable element; TF – Transcription factor; TGS – Transcriptional gene silencing; Thr – Threonine; TTS – Transcription termination site; Tyr – Tyrosine. *in balance with DNA demethylation mediated by ROS1.
Histone variants
The canonical histones H2A, H2B, H3, and H4, which are part of the nucleosome histone octamer, and the histone H1, which additionally stabilises it, can show variations in their primary structure. Different "subtypes" of the individual histones are referred to as histone variants. While histones H2A, H2B, H3, and H4 are expressed in the S-phase of the cell cycle and bind to the DNA molecule after replication, histone variants are expressed throughout the cell cycle and can replace the basic histones in the nucleosome at any point in the cell cycle (Jiang and Berger 2017). Different histone variants contribute to the dynamic chromatin landscape and exert a significant influence on essential nuclear processes. In addition, each of these variants has a different effect on the regulation of gene expression during development or stress response. Consequently, the abundance of histone variants has an immense potential to answer numerous unresolved questions in the field of eukaryotic epigenetics (Foroozani et al. 2022). The most important histone variants in plants (H2A.X, H2A.Z, H2A.W, H3.3, CenH3, H1.1, H1.2, and H1.3; Jiang and Berger 2017) are, together with their general functions, presented in Fig. 1 (orange). Sequences of expressed plant genes contain the canonical histone H2A and the histone variant H2A.X in their gene body (Lei and Berger 2020). The histone variant H2A.X plays a role in signalling damage to the DNA molecule that can occur due to light or oxidative stress (Nisa et al. 2019). Phosphorylation of H2A.X at serine residues is a signal that damage to the DNA molecule has occurred at a specific site (Lei and Berger 2020). The histone variant H2A.Z is predominantly found in euchromatic regions of the DNA molecule, either at transcription start sites of transcriptionally active genes or along the bodies of transcriptionally repressed genes (Sura et al. 2017, Lei and Berger 2020). The presence of H2A.Z in the first nucleosome downstream of the transcription start site (position +1) promotes transcription by preventing the stalling of RNA polymerase (Pol) II (Sura et al. 2017).
Histone variant H2A.W promotes chromatin condensation and is involved in the silencing of TEs in the pericentromeric heterochromatic region (Yelagandula et al. 2014). Although H2A.W colocalises with regions of the DNA molecule containing the histone modification H3K9me2 and cytosine methylation, the mechanism of deposition of H2A histone variant W in nucleosomes is independent of histone or DNA methylation processes (Yelagandula et al. 2014). The canonical histone H3, also known as histone variant H3.1, is predominantly located in the pericentromeric chromatin region where it is involved in silencing expression of TEs (Mito et al. 2005), and in the euchromatic regions of chromosomes (Jiang and Berger 2017). The histone chaperone Histone cell cycle regulator (HIRA) is involved in the deposition of the histone variant H3.3 instead of H3.1 in nucleosomes (Nie et al. 2014). The H3.3 variant predominates in nucleosomes of actively transcribed genes (Fig. 1, orange). Transcription termination sites contain the highest concentration of histone H3.3, which correlates with increased expression of these genes (Jiang and Berger 2017, Wollmann et al. 2017). Genes that contain H3.3 histone variant only in promoter regions are less strongly expressed and are subject to strong regulation (Jiang and Berger 2017). Interestingly, the presence of the H2A.Z histone variant in genes shows an inverse correlation with the presence of histone H3.3 (Nie et al. 2014, Jiang and Berger 2017). The CenH3 histone variant is located in the nucleosomes of the centromeric region of the chromosome and plays a key role in the formation of the kinetochore complex during cell division (Fig. 1, orange; Keçeli et al. 2020). Histone variants H1.1 and H1.2 are constitutively expressed in all cells and tightly bind part of the DNA molecule between the nucleosomes, preventing methyltransferases from accessing the DNA molecule (Jiang and Berger 2017). Under normal conditions the histone variant H1.3 is only expressed in the guard cells, where it is responsible for the correct opening of the stomata (Rutowicz et al. 2015). The expression of histone variant H1.3 together with H2A.Z is induced under conditions of water deprivation or after treatment with abscisic acid (ABA), when it is no longer restricted to the guard cells (Fig. 2A; Jiang and Berger 2017). Histone H1.3 is thought to cause hypermethylation of the DNA molecule because, unlike histone variants H1.1 and H1.2, it allows methyltransferases access to sequences that need to be methylated (Rutowicz et al. 2015, Jiang and Berger 2017).
Histone modification
The dominant histone modifications are methylation (me), acetylation (ac), phosphorylation, ubiquitination and SUMOylation (Fig. 1, orange). Since histone methylation marks play a crucial role in regulating chromatin structure and gene expression, changes in histone methylation are an important epigenetic regulatory factor in the response of plants to abiotic and biotic stress (Bobadilla and Berr 2016). Histones can be methylated at specific amino acid residues within their N-terminal tail domains. The most common methylated amino acid residues are lysine (K) and arginine (R). Histones can be mono-, di- and trimethylated on lysine residues, while on arginine residues they can be mono- or dimethylated (Liu et al. 2010). Histone H3 methylation marks such as H3K9me1, H3K9me2, and H3K27me1 are usually characteristic of silenced TEs in the heterochromatic regions of chromosomes (Chung et al. 2022). The histone modification H3K27me3 has a repressive effect on transcription in the euchromatic regions of chromosomes (Bobadilla and Berr 2016, Chung et al. 2022). The histone modifications H3R2me2 and H4R3me2, which are anticorrelated in their position with the modification H3K4me3, have a repressive effect on transcription (Bobadilla and Berr 2016). The histone modifications H3K4me3, H3K36me2, and H3K36me3 are located in the vicinity of actively expressed genes in the euchromatin region of the chromosome (Chung et al. 2022, Shi et al. 2023).
The targets of histone acetylation are specific lysine side branches at the N-terminus of histones H3 (K4, K9, K14, K18, K23, K27) and H4 (K5, K8, K12, K16, K20) (Liu et al. 2016). Like methylation, histone acetylation is involved in regulating the expression of transcription factors (TFs) involved in plant response to biotic and abiotic stress (Hu et al. 2019). In general, histone acetylation increases gene expression by promoting chromatin decondensation (Luo et al. 2017).
Protein kinases most frequently phosphorylate serine, threonine, and tyrosine residues of histones H3 and H2A.X. Phosphorylation of histone H3 plays a role in chromosome condensation, stimulation of gene expression, DNA damage repair (Chung et al. 2022), and response to stress caused by increased salinity, low temperature, or ABA treatment (Wang et al. 2015). Phosphorylation of the histone variant H2A.X, as previously mentioned, plays a role in DNA damage signalling (Lei and Berger 2020).
The monoubiquitination of histones in plants is a specific type of histone modification in which a single ubiquitin molecule binds with its C-terminus to a lysine residue in the histone. This post-translational modification occurs mainly in histones H2A and H2B (Feng and Shen 2014, Nunez-Vazquez et al. 2022). Monoubiquitination of histone H2A is associated with transcriptional repression controlled by Polycomb repressive complex 1 (PRC1), while monoubiquitination of histone H2B is associated with transcriptionally active loci (Chung et al. 2022) and is known to activate genes involved in plant responses to abiotic and biotic stress such as drought, salt, and fungal diseases (Fig. 2B; Dhawan et al. 2009, Chen et al. 2019, Sun et al. 2020).
The binding of small ubiquitin-like modifier (SUMO) proteins to lysine residues of histones is called SUMOylation and represents a post-translational regulation of protein function. The Arabidopsis proteome contains numerous proteins affected by SUMOylation, suggesting that this post-translational modification is as important as phosphorylation and ubiquitination (Augustine and Vierstra 2018). Many of the known SUMO substrates are nuclear proteins, including gene regulatory proteins, transcriptional coactivators/repressors, and chromatin modifiers, emphasising the crucial role of SUMOylation in the regulation of nuclear processes (Singh et al. 2022). An increased SUMOylation of nuclear proteins is a notable and rapid action in response to various stressors. However, the exact mechanism by which this modification contributes to stress resilience is still unclear. Several studies have reported the role of SUMOylation in altering gene expression in plant response to biotic and abiotic stress (Chen et al. 2011, Park et al. 2011, Ghimire et al. 2020). For example, Niu et al. (2019) observed that SUMOylation of Topless-related protein 1 (TPR1), a transcriptional co-repressor of immune responses in Arabidopsis, reduces its association with histone deacetylase 19 (HDA19) and decreases its activity in transcriptional co-repression, resulting in suppressed immunity.
Small non-coding RNAs
One of the greatest discoveries in RNA biology was the discovery of small non-coding RNAs (sncRNAs), which play a crucial role in various cellular processes. These sncRNAs, which are typically 20-30 nucleotides (nt) long, act at multiple levels, including chromatin remodelling and segregation, RNA processing and stability, transcription and translation. In addition, sncRNA molecules are actively involved in the process of DNA methylation known as RdDM (Borges and Martienssen 2015, Huang and Jin 2022). In plants, two main categories of sncRNAs are distinguished according to their origin and structure: microRNAs (miRNAs), which are usually 21-22 nt long (Axtell and Meyers 2018), and siRNAs, which are usually between 21 and 24 nt long (Borges and Martienssen 2015). MiRNAs, a subgroup of hairpin RNAs (Axtell 2013), are non-coding single-stranded RNA molecules that are involved in the process of post-transcriptional gene silencing (PTGS) in the cytoplasm (Meister 2013, Matzke and Mosher 2014). The second group of sncRNAs, known as siRNAs, is derived from double-stranded RNA precursors. Based on their function, siRNA molecules are divided into heterochromatic siRNAs (het-siRNAs), natural antisense transcript siRNAs (nat-siRNAs) and secondary siRNAs. While nat-siRNAs are involved in PTGS (Moldovan et al. 2010, Borges and Martienssen 2015), het-siRNAs and secondary siRNAs are involved in canonical RdDM (Ji and Chen 2012) and non-canonical RdDM (Borges and Martienssen 2015), respectively (Fig. 1, purple).
DNA methylation
DNA methylation is the addition of a methyl group from S-adenosylmethionine to the fifth carbon atom of cytosine, resulting in 5-methylcytosine (5-meC), catalysed by a family of methyltransferases (Fig. 1, green). Methylation in gene promoters generally leads to repression of nearby gene expression (Zhang et al. 2018), with some exceptions such as the promoter of the gene encoding the DNA demethylase Repressor of Silencing 1 (ROS1; Lei et al. 2015, Williams et al. 2015). In most cases, methylation prevents the binding of transcriptional activators and enhances the binding of transcriptional repressors (Kumar and Mohapatra 2021). In addition, methylation of promoter regions can influence the formation of post-translational modifications of histone tails that inhibit transcription (Zhang et al. 2018). Methylation in the gene body is a characteristic of constitutively expressed genes and occurs mainly within exons in the CG context that are far from the start and end site of transcription (Bewick and Schmitz 2017). It is hypothesised that the main role of methylation of the gene body is to prevent the binding of the histone variant H2A.Z, which influences gene expression depending on the developmental stage of the plant and environmental signals (Zilberman et al. 2008, Zhang et al. 2018). Intron methylation can influence the alternative processing of mRNA transcripts (Zhang et al. 2018).
During the life cycle of a plant, two types of DNA methylation alternate. One is responsible for the maintenance and inheritance of the established methylation state, and the other for de novo DNA methylation. The context of the sequence that will be methylated by a specific DNA methyltransferase depends on whether the role of that DNA methyltransferase is to preserve the existing DNA methylation marks or to methylate DNA de novo (Matzke and Mosher 2014). Methyltransferase 1 (MET1), a homologue of the mammalian protein DNA (cytosine-)-methyltransferase 1 (DNMT1), is responsible for the maintenance of CG methylation. Methyltransferase Chromomethylase 3 (CMT3), a homologue of mammalian DNMT3, is responsible for the maintenance of methylation in the CHG context. The third plant-specific methyltransferase, Domains rearranged methyltransferase 2 (DRM2), is the primary methyltransferase responsible for the CHH context, but also for the establishment of de novo methylation in all three contexts (He et al. 2011).
While methylation in symmetric contexts is inherited after DNA replication and can be passively maintained, the maintenance of methylation in an asymmetric CHH context is more complex and requires an active signal. In plants, this signal is mediated by siRNA molecules that direct the methyltransferase DRM2 to the target site and perform their function as part of the RdDM. RdDM is primarily involved in TEs silencing (Matzke and Mosher 2014, Zhang et al. 2018). TEs methylation reduces the expression of downstream genes (Hirsch and Springer 2017). The first step to control the activity of the newly inserted TEs is PTGS by activation of the non-canonical RdDM. Immediately after insertion, TEs are active and are transcribed by the enzyme Pol II. However, to counteract the potentially deleterious effects of TEs activity, some of the TEs RNA transcripts are recognised and bound by RNA-dependent RNA polymerase 6 (RDR6) that converts these single-stranded RNA molecules into double-stranded RNAs, which are then processed into 21-22 nt long siRNA molecules by the proteins Dicer-like 2 (DCL2) and Dicer-like 4 (DCL4). These siRNAs form a complex with the Argonaute 1 (AGO1) protein, which targets the complementary transposon transcripts for cleavage and degradation (Matzke and Mosher 2014). In addition to AGO1, siRNAs can also interact with Argonaute 2 (AGO2), the methyltransferase DRM2, Pol V and the protein Needed for RDR2-independent DNA methylation (NERD; Pontier et al. 2012). This interaction leads to DNA methylation being initiated de novo, which serves as a signal for the canonical pathway (Nuthikattu et al. 2013).
Canonical RdDM begins with the recruitment of Pol IV to specific target sites in the genome, primarily regions containing TEs. Once Pol IV is recruited, it first produces single-stranded P4-RNA, also known as Pol IV-dependent RNA, with the help of the chromatin-remodelling factor Classy 1 (CLSY1; Law et al., 2011). These single-stranded RNA molecules are then converted into long double-stranded RNAs by the activity of RNA-dependent RNA polymerase 2 (RDR2). In the next phase, the dsRNAs are processed into siRNAs, which are about 24 nt long. This processing is mediated by the protein Dicer-like 3 (DCL3). The small RNAs are then methylated at their 3' end (Ji and Chen 2012) and transferred to the cytoplasm before being incorporated into the Argonaute 4 (AGO4) protein, forming the RNA-induced silencing complex (RISC). As soon as they are loaded into AGO4, they are directed back into the cell nucleus. The second part of the mechanism begins with the activity of Pol V, which produces long non-coding RNAs. Once Pol V is recruited, it interacts with AGO4 via the C-terminal domain of its largest subunit – Nuclear RNA polymerase E1 (NRPE1). Due to their complementarity, the long non-coding RNA transcripts produced by Pol V pair with siRNA molecules from the RISC. Furthermore, the protein RNA-directed DNA methylation 1 (RDM1) binds AGO4 to methyltransferase DRM2 (Matzke and Mosher 2014).
In addition to methylation, demethylation of the DNA molecule is an important mechanism in the regulation of gene expression, TEs activity and the plant's response to stress. Passive demethylation occurs during replication of the DNA molecule by inactivating or reducing the concentration of enzymes involved in the maintenance of methylation (Li et al. 2018). Active demethylation occurs through the activity of DNA-glycosylases – ROS1, Demeter (DME) and DEMETER-like protein 2 and 3 (DML2/3), which actively cleave the methyl group from 5-meC (Li et al. 2018). This demethylation of TEs prevents the spread of methylation marks to nearby protein-coding genes and thus preserves the integrity of gene expression patterns (Tang et al. 2016). Moreover, an increased level of DNA methylation promotes the expression of ROS1 and counteracts the effect of RdDM on a considerable number of loci (Lei et al. 2015, Williams et al. 2015, Zhang et al. 2018). For this reason, ROS1 plays an important role in the activation of TEs, and the induction of genes involved in the stress response. Interestingly, the methylation status of the ROS1 gene promoter regulates the expression of the ROS1 protein, creating a feedback loop (Yang et al. 2022).
Epigenetic response to temperature stress
When plants are exposed to temperatures that are 10 to 15 °C above the optimal temperature for their growth, this is referred to as heat stress (Wahid et al. 2007). At the cellular and molecular level, heat stress leads to damage of membrane and cytoskeletal proteins, as well as the accumulation of misfolded proteins and reduced enzyme activity. Such changes lead to alterations in the transcriptome, proteome, and metabolome, which in turn lead to reduced rates of photosynthesis and cellular respiration and an overall reduction in plant growth and development (Zhao et al. 2020, Chung et al. 2022). The initial response of plants to heat stress is usually a rapid and transient increase in the production of heat shock proteins (HSPs) and the induction of an antioxidant network to overcome the deleterious effects of ROS accumulation (Mittler et al. 2012, Hasanuzzaman et al. 2020). HSPs are considered heat-induced molecular chaperones, i.e., they help in the proper folding, stabilisation, and refolding of other proteins in the cell. Their main function is to protect cellular components and maintain cellular homeostasis under elevated temperature conditions.
At the epigenetic level, the response of plants to elevated temperatures is mainly controlled by histone dynamics and RdDM (Popova et al. 2013, Lämke et al. 2016, Yang et al. 2018). Some of the most important histone modifications involved in heat stress response are H3K4me3 and H3K9ac. These modifications are known as active or permissive marks and are associated with open chromatin, making the underlying genes more accessible to the transcription machinery. Indeed, in heat-treated Arabidopsis, H3K4me3 and H3K9ac were strongly induced in the 5'-region of genes encoding the heat shock proteins HSP18.2, HSP22.0 and Ascorbate peroxidase 2 (APX2) and were associated with hyperinduction of these genes upon heat stress (Lämke et al. 2016). In addition, histone deacetylases 6 and 2C (HDA6/HD2C) are involved in the plant response to heat stress. Heat stress induces the expression of HD2C (Buszewicz et al. 2016), which interacts with other histone deacetylases, HDA6 and HDA19, as well as with methyltransferases and chromatin remodelling complexes (CRCs) from the SWItch/Sucrose Non-Fermentable (SWI/SNF) subfamily. This interaction suggests that histone deacetylation may be functionally linked to chromatin remodelling and DNA methylation, indicating that a complex regulatory network is involved in the response to heat stress (Chung et al. 2022). On the other hand, a histone acetyltransferase (HAT) called General control non-depressible 5 (GCN5) plays an important role in the generation of permissive histone modifications, such as H3K9ac and H3K14ac (Hu et al. 2015). These modifications occur in the promoter regions of certain genes, including HSFA3, which regulates the expression of HSPs (Fig. 2C; Schramm et al. 2007), and the gene encoding Ultraviolet hypersensitive 6 (UVH6), which is involved in DNA damage repair (Liu et al. 2003). Acetylation of histones in these promoter regions enhances gene expression and contributes to the plant's ability to cope with heat stress. In gcn5 mutants in which GCN5 activity is impaired, the expression of key genes such as UVH6, MBF1C and HSFA2/3 is reduced, resulting in lower thermotolerance (Hu et al. 2015). Studies on rice ( Oryza sativa L.) have shown that heat stress activates the expression of the protein Fertilization-Independent Endosperm1 (FIE1), which is part of the Polycomb repressive complex 2 (PRC2; Folsom et al. 2014, Miryeganeh 2021). When FIE1 is overexpressed, this leads to reduced seed size and premature cellularisation. Interestingly, the study also showed that the repressive histone modification H3K9me2 and methylation status are temperature sensitive. This suggests that the thermal sensitivity of seed enlargement may be influenced by changes in the epigenetic regulation of endosperm development during early embryogenesis.
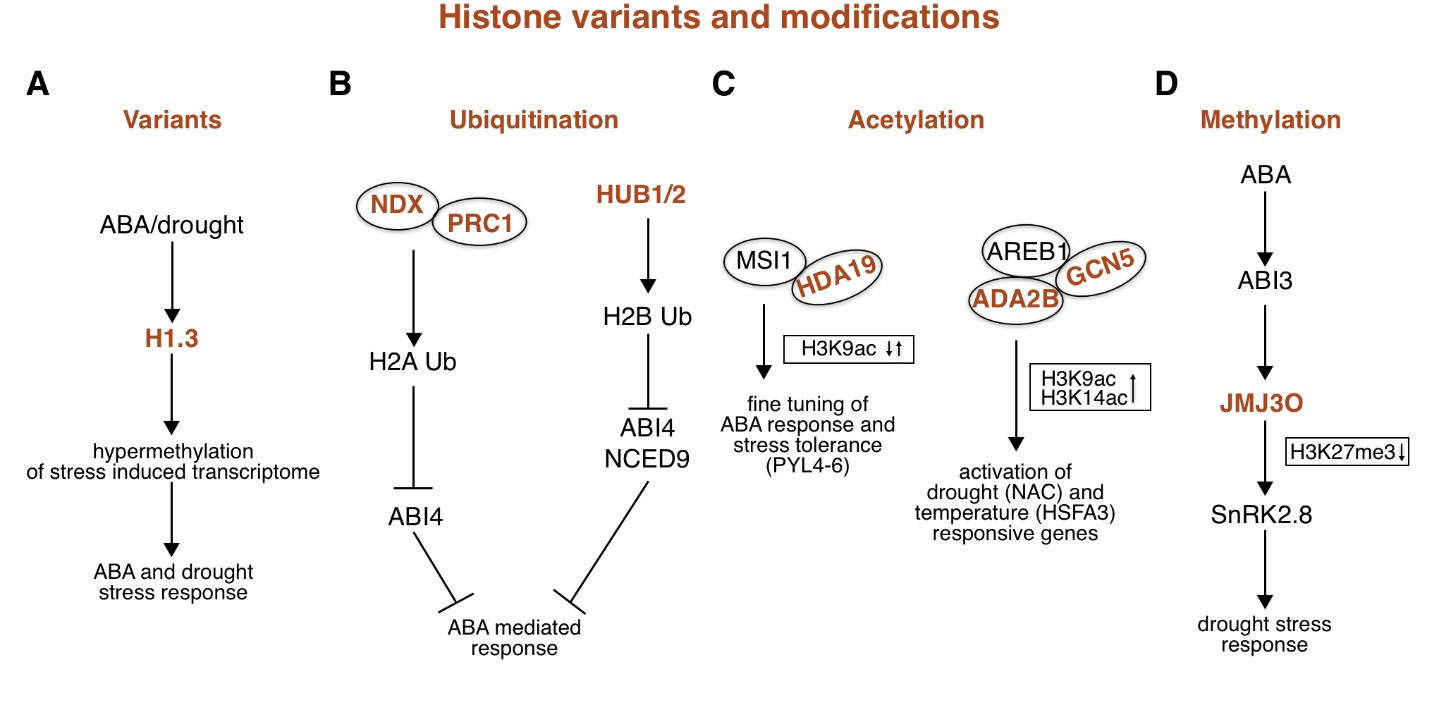
Fig. 2. ABA-related epigenetic role mediated by histone variants and modifications.
( A) Water deprivation and ABA treatment induce the expression of histone variant H1.3. In contrast to the tightly bound histone variants H1.1 and H1.2, H1.3 allows access to methyltransferases, resulting in hypermethylation of DNA loci involved in the response to ABA and drought stress. ( B) Under normal conditions (without ABA), NDX interacts with PRC1 and binds to the downstream regions of certain ABA-responsive genes, including ABI4. This interaction promotes monoubiquitination of H2A and thus represses the expression of ABA-responsive genes (left). HUB1/2 repress the ABA-signalling gene ABI4 and the ABA-biosynthesis gene NCED9 by H2B ubiquitination (right). ( C) The MSI1-HDA19 protein complex binds to the chromatin of ABA receptor genes, including PYL4‑ 6, and controls the fine-tuning of ABA signalling by maintaining a controlled level of histone H3K9ac mark (left). The AREB1-GCN5-ADA2b interaction complex binds to the ABRE motif of drought-responsive genes of the NAC family, where it increases H3K9ac levels and Pol II accumulation. GCN5 is also involved in controlling the expression of HSPs through H3K9 and H3K14 acetylation of temperature-responsive HSFA3 (right). ( D) ABA induces the expression of the transcription factor ABI3, which leads to the activation of the histone demethylase JMJ30. JMJ30 removes repressive histone modifications H3K27me3 from the promoter of SnRK2.8 kinase and promotes its expression, leading to plant response to abiotic stress and water deficit tolerance. Abbreviations: ABA – abscisic acid; ABI3/4 – ABA insensitive 3 and 4; ABRE – ABA-responsive elements; ADA2b – Alteration/deficiency in activation 2b; AREB1 – Abscisic acid‐responsive element binding protein 1; GCN5 – General control non-depressible 5; HDA19 – Histone deacetylase 19; HSFA3 – Heat shock factor 3; HSPs – Heat shock proteins; HUB1/2 – Histone monoubiquitination 1 and 2; MSI1 – Multicopy supressor of IRA1; NCED9 – 9-cis-epoxycarotenoid dioxygenase 9; NDX – Nodulin Homeobox Factor; Pol II – Polymerase II; PRC1 – Polycomb repressive complex 1; PYL4-6 – Pyr1-like 4-6; SnRK2.8 – SNF1-related protein kinase 2-8; ↑ – increase; ↓ – decrease.
In Arabidopsis, heat stress leads to globally increased methylation of the genome, while in other plants heat stress causes different trends and patterns of DNA methylation (Chung et al. 2022). However, the presence and proper function of the RdDM pathway is crucial for heat stress tolerance in plants, as mutants with non-functional canonical RdDM show a reduced response to heat stress (Popova et al. 2013). Under normal growth conditions, the Suppressor of drm1 drm2 cmt3 (SDC) protein is silenced by RdDM (Miryeganeh 2021). Heat stress activates the expression of SDC, leading to the activation of genes involved in the response to long-term heat stress and recovery. In other words, this activation suggests that the transcriptional response to heat stress must counteract the silencing effect of RdDM at specific genomic sites (Sanchez and Paszkowski 2014, Chang et al. 2020).
The RdDM pathway has also been shown to act as a protection against the activation and transposition of TEs during heat stress by maintaining their silencing and preventing possible genomic instability. Heat stress leads to the activation of certain TEs, such as the retrotransposon ONSEN in Arabidopsis, and mutants impaired in siRNA biogenesis show increased accumulation of ONSEN transcripts (Ito et al. 2011). In addition, ONSEN transposition has been detected in the progeny of heat-treated Arabidopsis with a non-functional RdDM pathway (Cavrak et al. 2014).
Two types of low-temperature stress can be distinguished: cold stress, which occurs at temperatures between 0 °C and 20 °C, and freezing stress, which occurs below 0 °C (Ritonga and Chen 2020). Cold stress triggers a series of complex changes in plant cells that affect photosynthesis, cellular respiration, water relations, mineral supply, and various metabolic activities (Guo et al. 2018, Ritonga and Chen 2020). The first response to cold stress occurs at the plasma membrane, where low temperatures can lead to reduced fluidity, conformational changes in membrane proteins and altered metabolite accumulation, which in turn results in changes in the redox state of the cell (Plieth et al. 1999, Orvar et al. 2000). As a result, receptors for Ca2+ ions, ROS and phytohormones are activated, and protein kinase cascades that regulate gene expression to increase cold tolerance are initiated (Theocharis et al. 2012, Guo et al. 2018, Guo et al. 2019). At temperatures below 0 °C, ice crystals form in the apoplastic solution, leading to a reduction in water potential and causing water to flow out of the cells, resulting in cell dehydration. The formation of ice crystals can lead to electrolyte leakage and changes in membrane lipids, resulting in further stress (Ritonga and Chen 2020). To survive, plants utilise cold acclimation, in which they accumulate cold-protective polypeptides (e.g., the Cold-regulated 15A protein) and osmolytes (e.g., soluble sugars and proline) to prevent intracellular ice crystal formation and cell damage (Ritonga and Chen 2020, Hassan et al. 2021).
C-repeat binding factors (CBF) are TFs that belong to the family of Dehydration response element binding factors (DREB; Medina et al. 1999) and are the most important regulators of plant responses to cold stress. They activate Cold regulated ( COR) genes, which are responsible for plant tolerance to low temperatures (Chang et al. 2020). ABA is also involved in this mechanism by inducing the expression of both CBF and COR genes, further enhancing the plant's ability to cope with low temperatures (Kidokoro et al. 2021).
The chromatin-remodelling factor Pickle (PKL) is involved in the CBF-dependent stress response in Arabidopsis. The pkl mutants have reduced expression of C-repeat binding factor 3 ( CBF3) and COR genes and show hypersensitivity to low temperatures (Yang et al. 2019). In their previous study, Yang et al. (2017) observed that pkl mutants have reduced methylation at a large number of RdDM target loci, but only a small number of these loci are responsible for controlling TEs activity. It has been proposed that PKL plays a role in shaping the chromatin landscape in RdDM target regions and thus acts as a chromatin remodelling factor that supports RdDM function (Yang et al. 2017). Interestingly, PKL in cooperation with Photoperiod independent early flowering 1 (PIE1), which belongs to the SWI2/SNF2-Related1 (SWR1) family of CRCs responsible for the exchange of H2A-H2B to H2A.Z-H2B dimer (Jiang and Berger 2017), and with the H3K27me3 methyltransferase Curly leaf (CLF) in the PRC2 complex also facilitates deposition of the repressive histone modification H3K27me3 near genes marked with H2A.Z histone variants (Carter et al. 2018).
It is noteworthy that cold stress reduces the deposition of H3K27me3 in the promoters of two COR genes, COR15A and Galactinol synthase 3 ( GOLS3), even after a return to normal growth temperature, indicating the role of H3K27me3 as a memory marker for recent transcriptional activity in Arabidopsis (Kwon et al. 2009). Consequently, PKL could influence the plant response to cold stress by supporting the functionality of the RdDM pathway and modulating the chromatin state of COR genes through H3K27me3-dependent mechanisms.
In addition to histone methylation, several other histone modifications play an important role in the response of plants to cold stress. The exposure of maize ( Zea mays L.) to low temperatures leads to an increased expression of histone deacetylases (HDACs) and thus to a reduced acetylation of repetitive sequences in the heterochromatic regions of the genome. The increased acetylation is associated with reduced DNA methylation and the reduced number of repressive H3K9me2 histone modifications in this part of the genome. However, when the plants are exposed to prolonged stress, methylation patterns are re-established (Hu et al. 2012). In Arabidopsis, overexpression of Histone deacetylase 2D (HD2D) increases the plant's tolerance to low temperatures. Another histone deacetylase, HD2C, is degraded after exposure to stress by the protein High expression of osmotically responsive genes 15 (HOS15), which is part of the E3-ubiquitin ligase complex. The proteasomal degradation of the ubiquitinated deacetylase HD2C leads to higher acetylation levels in the promoters of the COR genes, resulting in their activation. In addition, HOS15 facilitates the recruitment of the CBF TFs to the promoters of the COR genes, which further increases their expression (Chang et al. 2020). It appears that the balance between histone acetylation and deacetylation is crucial for the fine-tuning of gene expression during plant response to cold stress. Some genes involved in cold tolerance may need to be activated, while others need to be repressed to maintain cellular function. The interplay of acetylation and deacetylation allows plants to regulate the expression of specific genes in response to changing environmental conditions, facilitating their adaptation and survival under cold stress.
Several studies have demonstrated a positive correlation between upregulated anthocyanin biosynthesis and tolerance to low temperatures (Ahmed et al. 2015, Li et al. 2015, Xiang et al. 2021). A recent study by Sicilia et al. (2020) reported for the first time that genes involved in anthocyanin biosynthesis exhibit different levels of DNA methylation in response to cold stress. One of these genes is Ruby, which is regulated by a retrotransposon element. In the study by Sicilia et al. (2020), oranges with higher anthocyanin content were found to have lower DNA methylation levels in the promoter region of the Ruby gene after exposure to low temperatures. In other words, under conditions of low methylation, the activity of the retrotransposon increases, leading to higher expression of the Ruby gene and consequently higher anthocyanin content. These results provide valuable insights into how anthocyanin biosynthesis is epigenetically regulated in response to cold stress, which could contribute to the improvement of cold tolerance in plants.
Epigenetic control of drought and water deficiency
Water is an essential element for the growth and survival of plants. Its deficiency has negative effects on various plant processes, such as seed germination, photosynthesis, transpiration, and metabolite transport (Hao et al. 2019, Fan et al. 2020). Insufficient water availability or an increase in the osmotic potential in the soil poses a challenge for plant water uptake. Therefore, when plants perceive water deficiency, they undergo complicated reprogramming at multiple levels, including epigenetic, transcriptional, post-transcriptional and post-translational processes (Shinozaki and Yamaguchi-Shinozaki 2007, Manna et al. 2021). Recent studies have highlighted two key factors that contribute to this reprogramming. First, induction of gene expression in the ABA biosynthetic pathway, leading to an increase in ABA levels and expression of ABA-dependent genes (Osakabe et al. 2014). Second, widespread transcriptional changes in the expression of numerous genes that are not under the regulatory control of ABA but are associated with changes in chromatin organisation and structure (Khan and Zinta 2016).
In the early 2000s, a study showed that drought stress leads to increased expression of the enzyme 9-cis-epoxycarotenoid dioxygenase 3 (NCED3), which plays a crucial role in the biosynthesis of the hormone ABA (Iuchi et al. 2001). This increased expression is driven by the accumulation of H3K4me3 in the gene body. In another study by Ding et al. (2011), a protein called Arabidopsis trithorax 1 (ATX1) was found to be responsible for the accumulation of H3K4me3 modification and to play a role in both ABA-dependent and ABA-independent signalling pathways related to dehydration stress. In addition, genes encoding Responsive to desiccation 29A (RD29A), Low-temperature-induced 65 (LTI65), Responsive to desiccation 22 (RD22) and Related to AP2 4 (RAP2.4) undergo activating histone modifications such as H3K4me3 and H3K9ac in their promoter regions when induced under water-deficit conditions (Chang et al. 2020). The number of these histone modifications increases the longer and more intense the stress is. However, in the recovery period after the stress, the above-mentioned histone modifications are removed again (Chang et al. 2020). Under normal growth conditions, the repressive histone modification H3K27me3 is regulated by the polycomb group protein complexes, in particular PRC1 and PRC2. A study by Ramirez-Prado et al. (2019) investigated a PRC1-like protein in Arabidopsis, termed Like heterochromatin protein-1 (LHP1), and its involvement in the repression of MYC2 TF, a master regulator of the interplay between the ABA, jasmonic acid and ethylene response pathways. Loss of LHP1 resulted in reduced H3K27me3 levels in the gene bodies of NAC domain containing proteins 19 and 55 (ANAC019/055), which are part of the transcriptomic network downstream of MYC2. These two TFs have been characterised as positive regulators of drought tolerance and their upregulation increases tolerance to this environmental stress (Tran et al. 2004). Indeed, the lhp1 mutant showed phenotypes similar to the MYC2- and ANAC019/055-overexpressing plants, including increased resistance to aphids, sensitivity to ABA and improved drought tolerance (Ramirez-Prado et al. 2019).
In addition to methylation, drought stress often leads to changes in histone acetylation of drought-responsive genes, resulting in widespread changes in histone acetylation patterns throughout the plant genome. Histone acetylation is dynamically regulated by HATs and HDACs. Under drought stress, the expression of certain HAT genes, such as TaHAG2, TaHAG3 and TaHAC2, was upregulated in a drought-resistant wheat variety compared to other drought-sensitive varieties, suggesting that HAT genes play a role in improving drought tolerance (Li et al. 2022). The gene GCN5, which belongs to the HAT family, is a widely recognised enzymatic factor responsible for the acetylation of lysine residues at histones H3 and H4 (Gan et al. 2021). Li et al. (2019) showed that drought stress in Populus trichocarpa induces changes in histone acetylation at the whole genome level. H3K9ac mark was associated with upregulated genes, while downregulated genes showed reduced H3K9ac levels. Analysis of differentially expressed genes revealed a significant enrichment of the ABRE motif, which corresponds to the ABA-responsive element binding protein (AREB1), within H3K9ac-associated promoters (Fujita et al. 2005). AREB1 has been shown to interact with the HAT complex, which consists of GCN5 and the transcriptional adaptor Alteration/deficiency in activation 2b (ADA2b). This interaction leads to the recruitment of the complex to drought-responsive genes, including the NAC gene family, by binding to ABRE motifs. As a result, there is an increase in H3K9ac and the accumulation of Pol II, leading to the activation of drought-responsive genes (Fig. 2C). Interestingly, this process enables P. trichocarpa to effectively cope with drought stress and thrive. As for HAT genes, several studies reported that drought stress affects the expression of HDAC genes, resulting in altered histone acetylation in drought-related genes. However, the specific expression pattern was not reported. For example, in soybean ( Glycine max (L.) Merr.), drought treatment decreased the expression of several HDACs (Yang et al. 2018), while in rice ( O. sativa L.), certain HDAC genes were upregulated (Hou et al. 2021). In Arabidopsis, hda19 mutants exhibited increased drought resistance (Ueda et al. 2018), while overexpression of the HDAC gene HD2D increased tolerance to abiotic stresses, including drought (Han et al. 2016). Studies on the response of HDACs to drought stress have shed light on specific mechanisms involving HDA6 in Arabidopsis. HDA6 has been proposed to act as an ON/OFF switch for a complex drought-responsive metabolic pathway that leads to a metabolic switch from glycolysis to acetate synthesis, thereby stimulating the jasmonic acid signalling pathway for drought tolerance (Kim et al. 2017). Under normal conditions, HDA6 suppresses the expression of key enzymes of the acetic acid biosynthetic pathway by reducing the acetylation of histone H4. However, under drought conditions, HDA6 dissociates from these genes, allowing increased H4 acetylation and upregulation of transcription. This initiates the pathway of acetic acid biosynthesis, which ultimately increases drought tolerance through the accumulation of acetate (Kim et al. 2017).
DNA methylation is another important factor in the response of plants to water deficiency. Correlation analyses have shown that DNA methylation has differential effects on gene expression under drought stress, suggesting that it is involved in multiple regulatory pathways that directly or indirectly affect gene expression (Sun et al. 2022). Stress increases genome methylation of P. trichocarpa compared to wild-type plants, which in turn regulates the expression of numerous TFs. Methylation of genes encoding TFs generally increases their expression, while reduced methylation decreases the transcription rate (Liang et al. 2014). In rice ( O. sativa L.), it has been observed that cultivars sensitive to water deficit are hypomethylated, while those resistant to drought are hypermethylated (Gayacharan and Joel 2013). TEs are also subject to changes in DNA methylation caused by abiotic stress, including drought. In P. trichocarpa, about 65% of transposons located in the promoters of TFs were hypomethylated in response to water deficit, and the remaining 35% were hypermethylated (Liang et al. 2014). In maize ( Z. mays L.), transposons were discovered to regulate 33% of genes involved in the response to abiotic stress, and this regulation is not always stimulatory (Makarevitch et al. 2015). One example is the maize gene NAC111, where the insertion of miniature inverted-repeat transposable elements (MITEs) in the promoter region correlates with lower gene expression, leading to increased drought sensitivity (Mao et al. 2015). Interestingly, the same study showed that MITEs, when heterologously expressed in Arabidopsis, suppress the expression of ZmNAC111 via the RdDM pathway and the deposition of the repressive histone modification H3K9me2.
It is assumed that the SNF2/Brahma‐type protein Chromatin remodelling 12 (CHR12) is responsible for the temporary interruption of plant growth after drought and heat stress. A study by Mlynárová et al. (2007) showed that overexpression of CHR12 in Arabidopsis leads to stunted growth of typically active primary buds and diminished growth of the primary stem, particularly under stress conditions. In contrast, the CHR12 knockout mutant shows less growth inhibition when exposed to moderate stress than the wild type. This result is particularly interesting when considering another study by Han et al. (2012), which showed that loss of function of the SWI2/SNF2 chromatin remodelling ATPase Brahma (BRM) leads to increased drought tolerance, suggesting that BRM plays a crucial role in balancing growth and stress responses in plants.
ABA is a central mediator of the epigenetic code of climate-related abiotic stress in plants
Since the stressors associated with climate change (extreme temperatures, water deficiency, drought, and soil salinisation) often occur simultaneously in the context of climate change, the most effective responses of plant organisms should overlap or even be shared. The best-known common mediator of plant responses to water deficit, drought, extreme temperatures, and soil salinity is ABA (Vishwakarma et al. 2017). The ABA signalling pathway is activated by the recognition of phytohormone molecules by cell receptors, which initiate downstream signalling cascades that trigger various physiological effects (Wang and Zhang 2008). Briefly, ABA binds to Pyrabactin resistance 1, Pyr1-like and Regulatory components of ABA receptors (PYR1/PYL/RCAR) due to accumulation induced by stress signalling, resulting in inhibition of Protein phosphatases type 2C (PP2Cs). This inhibition in turn releases Sucrose nonfermenting 1-related protein kinases 2 (SnRK2s) and activates them by autophosphorylation. Once activated, SnRK2s play an important role in a number of vital biological processes, such as transcriptional regulation, RNA processing and epigenetic modifications (Wang et al. 2013b, Chang et al. 2020, Fidler et al. 2022).
Over the last twenty years, considerable efforts have been made to unravel the epigenetic mechanisms underlying ABA responses (Bulgakov et al. 2019, Chang et al. 2020). In the plant kingdom, there is a unique subset of minor H1 variants that respond to both drought and ABA. This subset, known as H1.3, is thought to play an important role in facilitating adaptive responses to stressful environmental conditions (Jiang and Berger 2017). Extensive studies of h1.3 null mutants have shown that the presence of H1.3 is essential for two important aspects: maintaining optimal stomatal functionality under normal growth conditions and enabling adaptive developmental responses when plants encounter the dual challenge of reduced light availability and water deficiency (Rutowicz et al. 2015). H1.3 may play a role in orchestrating changes in the stress-induced transcriptome by participating in DNA hypermethylation processes (Fig. 2A). This hypothesis is supported by the discovery that stress-induced DNA hypermethylation is significantly reduced in h1.3 mutants (Rutowicz et al. 2015). It has already been established that both H1.1 and H1.2 inhibit the interaction of DNA methyltransferases with DNA molecules (Zemach et al. 2013). Under stress conditions, H1.3 could compete with histone variants H1.1 and H1.2 that modulate DNA accessibility to DNA methyltransferases and associated epigenetic changes (Rutowicz et al. 2015).
New findings indicate that histone modifications are also involved in the ABA-mediated response to stress conditions. Histone ubiquitination regulates many genes associated with seed dormancy. The absence of the E3 ligase enzymes Histone monoubiquitination 1 and 2 (HUB1/2) reduces seed dormancy and represses genes such as 9-cis-epoxycarotenoid dioxygenase 9 ( NCED9) and ABA insensitive 4 ( ABI4; Chang et al. 2020). In Arabidopsis, Nodulin Homeobox Factor (NDX) interacts with components of the PRC1 complex, and together they lead to the silencing of ABA-dependent genes, such as ABI4, through histone H2A monoubiquitination (Zhu et al. 2020). In turn, ABA reduces the expression of the NDX protein and enables the activation of ABI4 gene expression, but it is also involved in the regulation of histone demethylation (Fig. 2B; Zhu et al. 2020).
A study by Mehdi et al. (2016) found that the WD40 repeat-containing protein Multicopy suppressor of IRA1 (MSI1) in Arabidopsis forms a complex with HDA19, a histone deacetylase. This MSI1-HDA19 complex controls the fine-tuning of ABA signalling transduction. It exerts its influence by binding to the chromatin of ABA receptor genes and maintaining a controlled level of histone H3 acetylation at lysine 9. This delicate balance ultimately influences ABA receptor gene expression (Fig. 2C). Interestingly, decreased levels of MSI1 or HDA19 were associated with increased salt stress tolerance, suggesting increased sensitivity of the genes to ABA. In addition, the complex was observed to target the promoters of the key ABA receptor genes, namely PYL4, PYL5 and PYL6, leading to repression of their expression (Mehdi et al. 2016). Histone deacetylases HDA6 and HD2C are involved in regulating the expression of two ABA-responsive genes, ABA insensitive 1 and 2 ( ABI1/ 2; Luo et al. 2012). These genes encode two phosphatases from the PP2C protein family, which negatively regulates the ABA response (Merlot et al. 2001). Indeed, hda6 and hd2c mutants were found to have increased ABI1/ 2 gene expression and lower resistance to aqueous NaCl and ABA treatment (Luo et al. 2012). Furthermore, histone methylation affects the expression of the SNF1-related protein kinase 2-8( SnRK2.8) gene, as treatment with ABA induces the expression of the ABA insensitive 3 ( ABI3) gene, leading to the activation of histone demethylase JMJ30. Subsequently, JMJ30 removes the repressive histone modifications H3K27me3 from the promoter of the SnRK2.8 gene and promotes its expression (Wu et al. 2019, Chang et al. 2020, Shi et al. 2023). This regulatory cascade is particularly important for the response of young seedlings to water deficiency (Fig. 2D; Wu et al. 2019).
It has been shown that after treatment with ABA, the chromatin structure undergoes CRC-mediated changes. The Switch/sucrose nonfermenting 3B (SWI3B) subunit of SWI/SNF CRCs interacts with Hypersensitive to ABA1 (HAB1), a member of the PP2C phosphatase family (Fig. 3A).
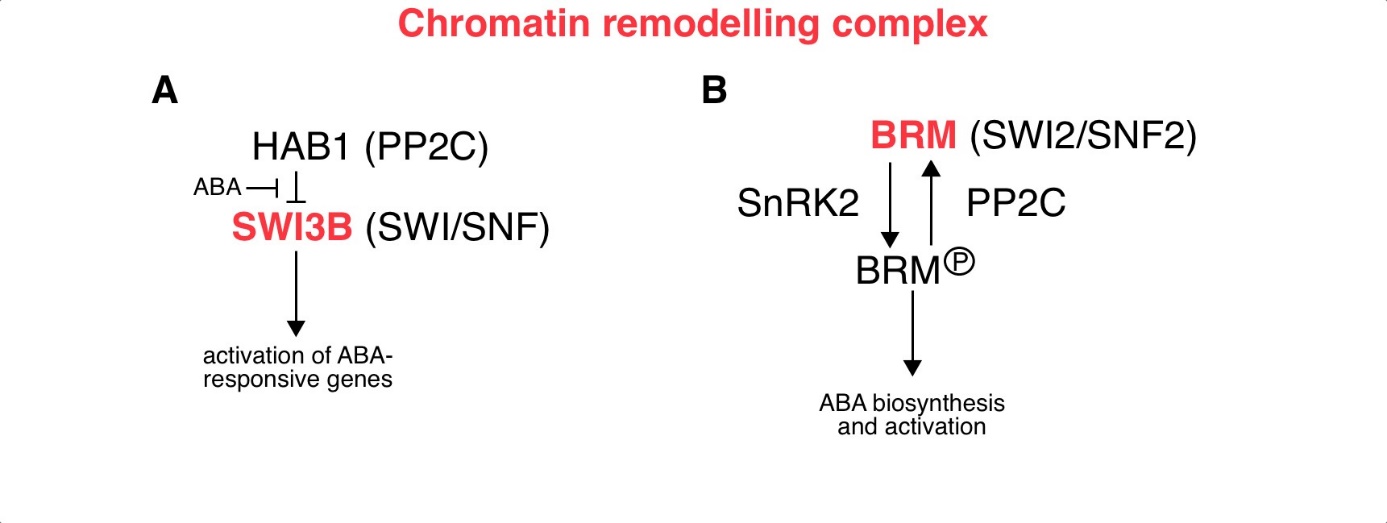
Fig. 3. ABA-related epigenetic role mediated by the activity of the chromatin remodelling complex.
( A) The PP2C phosphatase HAB1 mediates the inhibition of SWI3B, the subunit of SWI/SNF CRC, and thus prevents the expression of ABA-responsive genes. Under stress conditions, ABA accumulation neutralises the effect of HAB1 and allows SWI3B to activate the expression of ABA-responsive genes. ( B) The activity of the BRM is regulated by the process of phosphorylation and dephosphorylation. SnRK2 kinase-mediated phosphorylation of BRM leads to the release of BRM-mediated repression of ABA-responsive genes. In contrast, PP2C-mediated dephosphorylation of BRM serves to maintain its repressive role in the ABA response. Abbreviations: BRM – SWI2/SNF2 chromatin remodelling ATPase Brahma; CRC – chromatin remodelling complex; HAB1 – Hypersensitive to ABA1; PP2C – Protein phosphatases type 2C; SnRK2 – Sucrose nonfermenting 1-related protein kinases 2; SWI/SNF – SWItch/Sucrose Non-Fermentable subfamily; SWI3B – Switch/sucrose nonfermenting 3B.
Under normal growth conditions, HAB1 inhibits SWI3B and thus prevents the expression of ABA-responsive genes. However, treatment with ABA counteracts the effect of HAB1 and allows SWI3B to activate the expression of ABA-responsive genes (Saez et al. 2008). Another CRC subunit, BRM from the SWI2/SNF2 family, suppresses ABA-responsive genes in the absence of stress (Fig. 3B; Han et al. 2012). The activity of BRM is regulated by the interaction of SnRK2 kinases and PP2C phosphatases. In the signalling pathway, BRM is located downstream of SnRK2s. Phosphorylation of BRM by SnRK2s leads to the release of BRM-mediated repression of ABA-responsive genes. Conversely, PP2C-mediated dephosphorylation of BRM maintains the repressive role of BRM in the response to ABA (Peirats-Llobet et al. 2016). SWR1 CRCs are involved in the exchange of H2A-H2B for the H2A.Z-H2B dimer in nucleosomes (Jiang and Berger 2017), while CRCs from the INO80 subfamily are involved in the reverse process (Fig. 3C; Han et al. 2015). The exchange of H2A.Z-H2B dimers is faster than that of H2A-H2B dimers (Brahma et al. 2017). As a result, genes covered by H2A.Z-H2B nucleosomes can respond faster to stimuli. Indeed, histone H2A.Z plays a role in the response of plants to osmotic stress and is removed from induced genes under conditions of water deficit (Sura et al. 2017). Under heat stress conditions Heat shock factor 1 (HSF1) TF is involved in the removal of histone H2A.Z from genes induced at a temperature of 27 °C (Cortijo et al. 2017).
ABA-mediated responses synergise with other regulatory mechanisms, including the plant-specific epigenetic pathway of RdDM. A study by Kim et al. (2019) has shown that the expression of several ABA-dependent genes is regulated by the RdDM pathway and ROS1-dependent DNA demethylation (Fig. 4A). In ros1 mutants treated with ABA, certain ABA-dependent genes are hypermethylated in a promoter region, leading to reduced gene expression. Examples of such genes are Nicotinamidase 3 ( NIC3), whose gene product is involved in the recycling of the cofactor NAD+ (Kim et al. 2019), and RD29A (Gong et al. 2002), which is involved in responses to abiotic stress. In addition, DNA methylation in response to ABA can influence the localisation of proteins in cells. RdDM and hd2c mutants show defective cellular localisation of two DEAD-box RNA helicases, Stress response suppressor 1 and 2 (STRS1/2), in response to abiotic stress, including treatment with ABA (Khan et al. 2014). Moreover, treatment with ABA reduces the expression of STRS1/2 helicases, while strs1/2 mutants exhibit increased tolerance to heat, water and salt stress, and upregulation of stress-responsive genes (Fig. 4B; Khan et al. 2014). A recent study on maize has shown that loss of Mediator of paramutation 1 (MOP1) protein activity leads to increased expression of ABA-responsive genes (Vendramin et al. 2020). Interestingly, MOP1 is an orthologue of Arabidopsis RDR2, a protein known for its interaction with Pol IV (Haag et al. 2014). This interaction is essential for Pol IV-dependent siRNA biogenesis and the functionality of the RdDM pathway (Fig. 4C). Madzima et al. (2021) discovered a list of genes that were uniquely up- and downregulated upon both ABA treatment and loss of MOP1. By comparing these unique genes to genes whose promoters share homology with MOP1-dependent siRNAs, the group found that a quarter of the genes are directly regulated by MOP1. In addition, a comprehensive Gene Ontology (GO) analysis revealed that there were more significantly enriched GO terms associated with upregulated genes than with downregulated genes. Since GO terms are associated with various biological processes, including signal transduction, cell communication, regulation of gene expression, and response to abiotic stimuli, the authors propose that MOP1-dependent activity and siRNAs are directly related to the regulation of certain biological processes in response to ABA. On the other hand, siRNAs associated with downregulated genes are probably independent of MOP1 and may have a less specific biological role associated with ABA responses (Madzima et al. 2021).
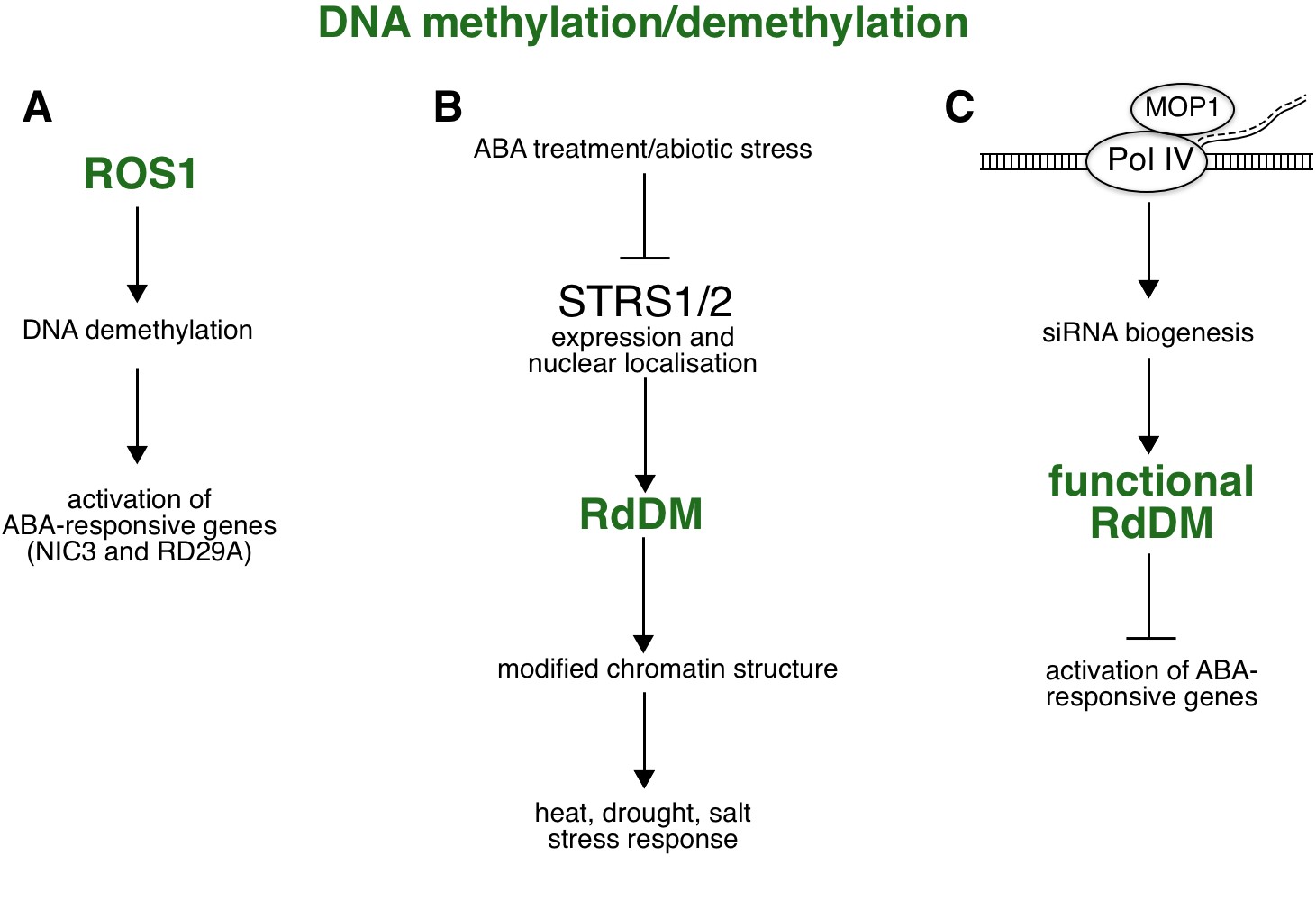
Fig. 4. ABA-related epigenetic role mediated by RdDM.
( A) Increased level of ROS1-mediated DNA methylation is required for expression of ABA- and stress-responsive genes such as RD29A and NIC3, suggesting a role for ROS1-dependent DNA demethylation in ABA-mediated stress responses. ( B) RdDM in response to ABA reduces the expression and affects the localisation of two DEAD-box RNA helicases, STRS1/2, thereby enhancing the expression of stress-responsive genes and increasing tolerance to heat, water, and salt stress. ( C) MOP1, the maize orthologue of Arabidopsis RDR2, represses expression of ABA-responsive genes via Pol IV-dependent siRNA biogenesis of RdDM. Abbreviations: MOP1 – Mediator of paramutation 1; NIC3 – Nicotinamidase 3; Pol IV – Polymerase IV; RD29A – Responsive to desiccation 29A; RdDM – RNA-directed DNA methylation; RDR2 – RNA-dependent RNA polymerase 2; ROS1 – Repressor of Silencing 1; siRNA – small interfering RNA; STRS1/2 – Stress response suppressor 1 and 2.
Conclusion and future perspectives
Numerous studies have shown that epigenetic processes, including DNA methylation, histone modifications and variants, and chromatin remodelling contribute to the response to stressors and enable plants to defend themselves and thrive under difficult conditions. These mechanisms also play a crucial role in hormonal signalling, regulation of antioxidant enzyme levels, and activation of stress resistance genes. Of particular interest is the fact that some epigenetic patterns can be inherited, increasing the adaptability of future generations to a stressful environment. Understanding the intricate regulatory processes involving epigenetic regulation as a target of stress and a force of adaptation can lead to the development of tools to protect crops from the growing challenges posed by pervasive stressors in global agriculture. ABA is involved in numerous aspects of stress responses, including almost all epigenetic mechanisms, and there are examples of overlap between the regulatory mechanisms of ABA and epigenetic adaptations to the environment. Therefore, elucidating the interplay between stressors, ABA and the epigenetic code could serve as a basis for the development of tools to optimise the synergistic responses of plants to global environmental change. Using the link between ABA-responsive element binding factors (ABFs) and ABA-responsive elements (ABREs) to make specific epigenetic marks at genetic loci and modulate gene expression is a promising way to improve breeding programmes. This approach enables precise and controlled adjustment of epigenetic modifications at key genetic loci and enables crops to develop a customised and efficient response to various stress factors. By strategically utilising this molecular partnership, researchers can contribute to the development of crops that are not only highly adaptable to adverse conditions, but also meet the growing demands of global agriculture in the face of changing environmental conditions. This advance has the potential to transform plant breeding and usher in an era of stress-resistant and high-yielding crop varieties.
Acknowledgements
The review was written in a support of the journal Acta Botanica Croatica on the occasion of its 100th anniversary.
We sincerely thank Željka Vidaković-Cifrek for the motivation of M.G., J.D. and S.V. to acquire knowledge about plant responses to stress.
Realization of this work was supported by grants from the Croatian Science Foundation (project PHYTOMETHDEV; IP 2016-06-6229 to DLL) and the University of Zagreb, Croatia.