
INTRODUCTION
Thiopurines were first discovered by Gertrude B. Elion and George H. Hitchings in the early 1950s (1). For their groundbreaking contributions to drug development, the pair was awarded the Nobel Prize in Medicine or Physiology in 1988. Since their discovery, thiopurines, including azathioprine (AZA), 6-mercaptopurine (6-MP), and 6-thioguanine (6-TG), have demonstrated their efficacy in various clinical applications. Thiopurines continue to be widely used in the treatment of chronic systemic autoimmune diseases, and prevention of transplant rejection, and remain an integral part of treatment for acute lymphoblastic leukemia (ALL) (2, 3).
Thiopurines are used to maintain remission, especially of corticosteroid-dependent or refractory inflammatory bowel disease (IBD) in adult patients (2). While the introduction of biologics has changed the therapeutic landscape of IBD, thiopurines continue to be an essential, efficient, and cost-effective component of IBD treatment, either as monotherapy or in combination with other treatments, particularly infliximab (2, 4–7). In the treatment protocol for childhood acute lymphoblastic leukemia (ALL), 6-MP serves as the backbone during the final and most prolonged phase of AIEOP BFM ALL 2017 protocol, the maintenance phase, which lasts up to two years (3). The continued relevance of thiopurines in clinical practice is further supported by their use in the treatment of other diseases such as acute myeloid leukemia, various lymphomas, rheumatoid arthritis, systemic lupus erythematosus, psoriasis, eczema, autoimmune hepatitis, idiopathic thrombocytopenia, and transplant medicine.
Thiopurines therefore remain an important anticancer and immunosuppressive group of drugs. However, they are characterized by a complex metabolism and narrow therapeutic index that exposes patients to the risk of severe, potentially life-threatening side-effects (8, 9). Due to individual genetic or metabolic differences, the same dose may achieve the desired effect in one patient while provoking a serious adverse reaction in another (10, 11). Therefore, precise dosing is an important factor to consider when using them. To address this challenge, pharmacogenomic guidelines for thiopurine S-methyltransferase (TPMT) genetic polymorphisms and TPMT activity have been in place for more than a decade (12), while recent advances in genomics also include the determination of the most common genetic polymorphisms in nudix hydrolase 15 (NUDT15) (11). In addition, therapeutic drug monitoring that allows quantification of thiopurine metabolites, especially 6-thioguanine nucleotides (6-TGN) and 6-methylmercaptopurine (6-MMP), is an important component in the optimization of thiopurine treatment if there is no response to thiopurines and/or in case of side-effects especially during IBD treatment (2, 13).
In this review, we highlight biomarkers for thiopurine therapy, namely, genetic polymorphisms in TPMT and NUDT15, and thiopurine metabolites. We first focus on the molecular basis of genotype-phenotype correlations and how these associations have led to well-established guidelines for thiopurine dosing. We then describe methods and clinically validated analytical procedures for TPMT and NUDT15 genotyping, measurement of TPMT activity, and monitoring of thiopurine metabolites. Finally, we address future challenges, particularly in relation to clinical implementation, and what are the approaches to overcome them.
THIOPURINES
Treatment with thiopurines lasts from one to several years, primarily during the maintenance phases of ALL and IBD, with the aim of extending disease remission. The most common starting daily dose of 6-MP in the maintenance phase of ALL treatment is 50 to 75 mg m–2 (1.5 to 2.5 mg kg–1), while IBD patients usually receive 1–3 mg kg–1 of AZA for maintaining remission (4, 14). The effectiveness of thiopurine treatment varies and depends on the type of disease and the age of patients. Due to a systematic approach to designing treatment protocols, the effectiveness in the field of ALL has increased from 40 % in the mid-1960s (15) to over 90 % in the pediatric population in recent years (3, 16). Nevertheless, some studies report only a 35 % effectiveness rate for thiopurine treatment in IBD (17, 18). Up to 60 % of patients discontinue treatment due to severe adverse effects, such as hypersensitivity reactions, acute pancreatitis, gastrointestinal toxicity, leukopenia, hepatotoxicity, and infections, or non-response to thiopurines (19, 20).
Metabolic pathways
Thiopurines are antitumor and immunosuppressive prodrugs, that undergo complex metabolism involving both activation and deactivation pathways (Fig. 1). After oral administration, the conversion of thiopurines begins in the intestines and liver, where AZA is partially enzymatically cleaved by glutathione S-transferase (GST) forming methyl nitrothioimidazole and 6-MP. The latter may enter the activation pathway resulting in the production of cytotoxic 6-TGN. This process starts with the attachment of ribosyl and phosphate groups to the purine moiety by hypoxanthine phosphoribosyltransferase (HPRT) and continues with an oxidative reaction catalyzed by inosine 5’-monophosphate dehydrogenase (IMPDH), which significantly influences the conversion rate to 6-TGN. The next step involves the conversion of the resulting 6-thioinosine monophosphate (TIMP) to 6-thioguanosine monophosphate (TGMP) by guanosine 5’-monophosphate synthase (GMPS). This is followed by phosphorylation by kinases to 6-thioguanosine triphosphate (TGTP) (9). This latter step is opposed by dephosphorylation with the NUDT15 (21).
Apart from the activation, there are two deactivation pathways of thiopurines, involving TPMT and xanthine oxidase (XO) (9). As a liver enzyme, XO converts between 3 and 37 % of absorbed 6-MP into thiouric acid (10). The remainder of 6-MP then either enters the activation pathway or is methylated by TPMT, resulting in the formation of 6-MMP. TPMT also participates in the methylation of some other 6-MP metabolites at various stages of the activation pathway (Fig. 1) (9). Namely, it is involved in the production of 6-methylthioinosine monophosphate (MTIMP), which, alongside 6-TGN, exhibits pharmacological effects (22).
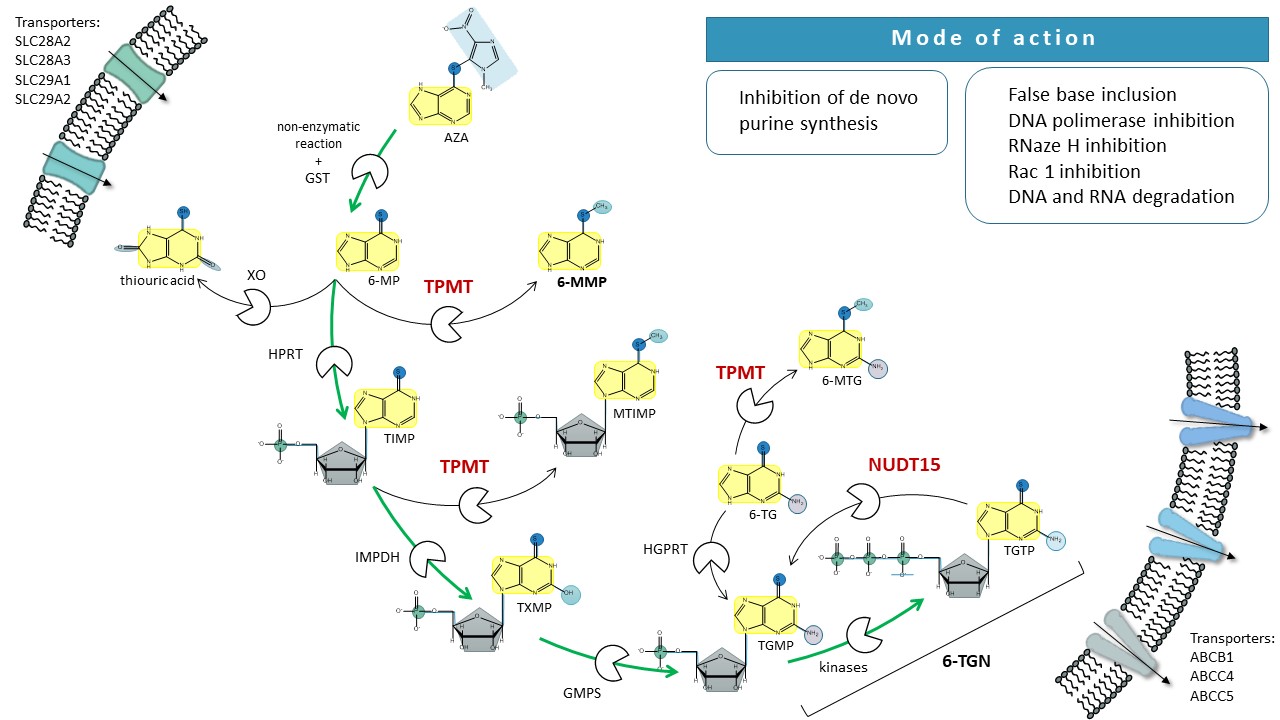
Fig. 1. Schematic presentation of thiopurine metabolism. 6-MP – 6-mercaptopurine, 6-MMP – 6-methylmercaptopurine, 6-MTG – 6-methyltioguanine, 6-TG – 6-tioguanine, 6-TGN – 6-tioguanine nucleotides, ABC – ATP-binding cassette (transporters), AZA – azathioprine, GMPS – guanosine monophosphate synthetase, GST – glutathione S-transferase, HPRT – hypoxanthine-guanine phosphoribosyltransferase, IMPDH – inosine-5′-monophosphate dehydrogenase, ITPA – inosine triphosphate pyrophosphatase, MTIMP – methylthioinosine monophosphate, NUDT15 – nudix hydrolase 15, XO – xanthine oxidase, SLC – solute carrier (transporters), TIMP – thioinosine monophosphate, TGMP – thioguanosine monophosphate, TPMT – thiopurine S-methyltransferase, TGTP – thioguanosine triphosphate, TXMP – thioxanthine monophosphate.
The activation and deactivation pathways for 6-TG are similar to the metabolism of 6-MP but are less complex since 6-TG, unlike 6-MP, already holds a guanine structure. Only a one-step conversion by HPRT transforms 6-TG into cytotoxic 6-TGN (Fig. 1) (9). The oxidative deactivation of 6-TG by XO has not been fully elucidated. 6-TG is also deactivated by TPMT methylation; however, its methylated metabolites do not demonstrate pharmacological activity (23).
Mode of action
Due to its structural similarity to guanine, 6-TGN incorporates as a building block into nucleic acids; however, instead of participating in synthesis, it disrupts the function of DNA polymerase, and thereby arrests replication of DNA, causing DNA degradation (24). The activity of 6-TGN includes the direct inhibition of DNA polymerase and ligase, as well as the suppression of RNase H activity, leading to a termination in gene transcription (25). The disintegration of nucleic acids triggers apoptotic mechanisms and cell death. The cytostatic and immunosuppressive effects of thiopurines, specifically TGTP, are also achieved through the obstruction of CD28-dependent activation of Rac1, which directs T lymphocytes toward apoptosis (26). Additional activity of thiopurines is expressed by MTIMP. By inhibiting phosphoribosyl pyrophosphate amidotransferase, it suppresses de novo purine synthesis and thereby reduces the level of adenosine triphosphate (22). Another target effect of thiopurines is triggered by methyl nitrothioimidazole, a degradation product of AZA. Its immunosuppressive effect is associated with the inhibition of oxidative stress defense mechanisms, as it can reduce the concentration of glutathione in the cell (27).
Safety and efficacy of treatment with thiopurines
Among patients treated with thiopurines, the most common motives for discontinuing therapy include hypersensitivity reactions, acute pancreatitis, gastrointestinal intolerance, myelotoxicity manifested as neutropenia, hepatotoxicity, and infections (28). Due to the potential mutagenic and carcinogenic activity of thiopurines, some patients who have successfully undergone treatment later reported developing secondary neoplasms, such as lymphoma, non-melanoma skin tumors, and chronic myeloid leukemia (29–31).
To ensure patient safety and efficacy of treatment with thiopurines, routine checks of the leukocyte levels and liver function through liver enzymes are mandatory during treatment, usually every three months in patients with IBD (32) and every 8–12 weeks during maintenance treatment of ALL (33). Where available, the level of thiopurine metabolites in hemolysates is frequently monitored in patients unresponsive to treatment or in individuals exhibiting side-effects (2, 13, 34–36). Levels of 6-TGN in the range of 235 to 450 pmol/8×108 red blood cells (RBC) in patients with IBD predict successful treatment (37). 6-TGN levels lower than 235 pmol/8×108 RBC are associated with non-responsiveness to therapy or indicate non-compliance (38, 39). Levels of 6-TGN higher than 450 pmol/8×108 RBC are associated with an increased risk of adverse effects (40, 41). Significantly elevated methylated thiopurine metabolites – 6-MMP > 5700 pmol/8×108 RBC, indicate a higher risk for liver toxicity, although monitoring 6-MMP alone is insufficient for predicting hepatotoxicity (4, 37, 42). A high ratio of 6-MMP to 6-TGN has proven useful, especially for detecting non-responders or shunters, a subgroup of patients, that excessively produce 6-MMP instead of 6-TGN.
Pharmacogenomic determinants of thiopurine metabolism
Alongside the nature of the disease, inherited predisposition significantly influences the acute onset of adverse effects, disease relapse, adverse effects that occur years after completion of therapy, and non-response to thiopurines. Genetic polymorphisms are one of the factors that can lead to insufficient efficacy and decreased safety of the drug when different individuals are exposed to the same dose (45). Clinical and translational studies have identified a number of polymorphisms affecting the clinical response to thiopurines, correlating with their efficacy, toxicity, or resistance to such treatment (8). However, only variants in TPMT and NUDT15 have been translated into clinical practice (11). We highlight these two genes and their implication in thiopurine therapy in the following sections.
Thiopurine S-methyltransferase
Thiopurine S-methyltransferase (TPMT) is a cytosolic enzyme that catalyzes the S-methylation of aromatic and heteroaromatic sulfhydryl compounds, with the methyl group being donated by the co-factor S-adenosyl methionine (SAM) (46, 47). The reaction, which presumably follows the classic nucleophilic substitution (SN2) mechanism (48), results in the formation of a methylated substrate and S-adenosyl homocysteine (SAH) (46). SAM serves not only as a methyl donor but also stabilizes TPMT, preserving its enzymatic activity, which was highlighted in our previous studies (49, 50). The gene encoding this protein is located on chromosome 6 at locus 6p22.3. The gene spans over 34 kb and consists of 9 exons, of which 8 code for the protein (51). In the human genome, pseudogenes for TPMT are located on chromosomes 18 and X (51).
Currently, 46 different single nucleotide polymorphisms (SNPs) as well as deletions of smaller gene fragments are known in the exonic or intronic region (52, 53). In addition to the SNPs, individuals also differ in the sequence of the TPMT promoter region with a G/C-rich variable number of tandem repeats (VNTRs), which are evolutionarily related to the most common SNPs (54). Due to their impact on enzyme activity and their significant frequency, the most important genetic polymorphisms are TPMT*3A, TPMT*3C, and TPMT*2 (55, 56, 57, 58). In these polymorphisms, a change in one nucleotide leads to a protein with an altered amino acid sequence, resulting in incorrect folding and faster degradation of TPMT (59). Consequently, the activity of TPMT corresponds to the individual's genotype, where individuals with wild-type TPMT have normal TPMT activity, heterozygous individuals have intermediate TPMT activity, and individuals carrying both variant alleles express TPMT with barely detectable enzymatic activity (58). In the European population, about 89 % of individuals have normal TPMT activity, 11 % have reduced TPMT activity, and around 0.3 % have very low TPMT activity (10, 58). However, the distribution of activities in individuals with the wild-type genotype is relatively wide, meaning that the genotype of the most common polymorphisms does not perfectly explain the phenotype and that there are other genetic or non-genetic factors that additionally influence TPMT activity (60).
Nudix hydrolase 15
Nudix hydrolase 15 (NUDT15) is an enzyme that belongs to the nudix hydrolase family of enzymes encoded by 24 genes on the human genome, which contains also at least 5 pseudogenes (61). These enzymes catalyze the hydrolysis of substrates with a general nucleoside diphosphate structure linked to another moiety, such as canonical and oxidized derivatives of (d)NTPs, nucleotide sugars, and dinucleotide polyphosphates. They share an evolutionary conserved domain named NUDIX box consisting of 23 amino acids, among which glutamic acid residues bind divalent cations, while other amino acids contribute to substrate specificity (61). In vitro studies have shown that NUDT15 preferentially hydrolyses 6-thio-deoxyguanosine triphosphate (6-TdGTP) and TGTP, the active metabolites of thiopurines, and negatively affects their desired cytotoxic and immunosuppressive effects, as well as increases the risk of thiopurine-related toxicities (62). Spanning over 15,030 base pairs on chromosome 13, the NUDT15 gene has 3 exons and corresponding intronic regions (63). The first association of NUDT15 variant (rs116855232) with thiopurine-induced toxicity came from a GWAS conducted in Korean patients with IBD (64) and was later confirmed in several other studies in patients with IBD and ALL (21, 65, 66). The identified missense variant c.415C>T, which results in a p.Arg139Cys change which leads to decreased enzyme activity, is present in NUDT15*2 and NUDT15*3 alleles. To date, several other variants have been identified and 20 different NUDT15 alleles together with additional 7 suballeles have been reported in the Pharmacogene variant consortium database (67). Based on in vitro activity data or in silico prediction, alleles are divided into those having a normal function (NUDT15*1 and it's 7 suballeles), alleles with severely decreased enzyme activity (NUDT15*2 - *9), alleles with no function (NUDT15*10, *13, *14, *17, *18 and *19) and alleles with unknown function (NUDT15*11, *12, *15, *16 and *20) (68).
Clinical implications and guidelines in thiopurine pharmacogenomics
Three TPMT alleles (TPMT*2, TPMT*3A, and TPMT*3C) constitute 90 % of cases of reduced activity phenotypes in individuals of European origin (10, 58, 69). As the presence of these alleles affects TPMT activity, poor TPMT metabolizers convert more thiopurines to the active 6-TGN, thus increasing the risk for drug toxicity, such as bone marrow suppression, severe gastric intolerance, and susceptibility to infections (70, 71). Similarly, individuals with genetic polymorphisms NUDT15*2, NUDT15*3, and NUDT15*9 exhibit lower NUDT15 activity and a higher risk for drug toxicity when treated with thiopurines (21, 69). Therefore, thiopurines require tailored dosing strategies based on TPMT and NUDT15 genetic variability. Genotyping of TPMT and NUDT15 prior to treatment can mitigate adverse thiopurine effects such as myelosuppression and leukopenia, without compromising the disease’s relapse rate (72–74).
International expert panels from the field of pharmacogenomics – Clinical Pharmacogenetics Implementation Consortium (CPIC), Dutch Pharmacogenetics Working Group (DPWG), and Canadian Pharmacogenomics Network for Drug Safety (CPNDS), have been established to consolidate information on how genetic polymorphisms impact treatment (11, 75, 76). These organizations have developed recommendations and pharmacogenomic guidelines for dosing of currently slightly less than 200 drugs, including thiopurines (75). Based on the functional impact of the variant alleles, minor allele frequencies in multiethnic populations, the availability of reference materials, and the feasibility for laboratories to test using standard methods, genetic polymorphisms in TPMT and NUDT15 can be classified into two Tiers (77). Tier 1 variant alleles include TPMT*2, TPMT*3A, TPMT*3B, TPMT*3C, and NUDT15*3, as they strongly affect enzyme activity, are tested by standard methods, and account for > 90 % or >50 % of the reported variant alleles, in TPMT or NUDT15, resp. Tier 2 includes other variant alleles in TPMT and NUDT15 that are less frequent with less defined molecular mechanisms or difficulty to test for them by standard methods (77).
According to the presence of Tier 1 genetic polymorphisms in TPMT in NUDT15, guidelines classify individuals as normal metabolizers with two functional (non-variant) alleles, intermediate metabolizers with one functional and one non-functional (variant) allele, and poor metabolizers with two non-functional (variant) alleles in respective genes. CPIC guidelines further classify individuals with one allele of uncertain function and one non-functional allele (i.e., TPMT*2/TPMT*8, TPMT*3A/TPMT*7, NUDT15*2/NUDT15*5, and NUDT*3/NUDT15*6) as possible intermediate metabolizers. According to guidelines (Table I), normal metabolizers should start with standard thiopurine doses, while intermediate metabolizers and possible intermediate metabolizers for either of the enzymes, should receive 30–80 % or 50–80 % of standard AZA/6-MP or 6-TG dose, resp. Poor metabolizers, diagnosed with malignancies, should start 6-MP at doses reduced by 10-fold thrice weekly, whereas in non-malignant cases it is suggested to consider alternative treatments to AZA. Alternative treatment is recommended also for poor metabolizers receiving 6-TG. In patients who are poor metabolizers due to the double variant NUDT15*3 genotype, 25 % of the 6-TG normal dose is recommended for malignancies. Additional guidelines refer to patients who are intermediate metabolizers for both enzymes (TPMT and NUDT15) (11, 78–80). Such patients are suggested to start with a 20–50 % reduction of the thiopurine standard dose at the beginning of the treatment, followed by potential further dose adjustments based on the severity of myelosuppression (81).
Guidelines issued by various working groups exhibit minor discrepancies. For instance, according to the DPWG guidelines, dose reduction is not required for intermediate metabolizers of 6-MP or AZA when they are used to achieve immunosuppression, provided the initial dose does not exceed 1.5 mg kg–1 for AZA or 0.75 mg kg–1 per day for 6-MP. Additionally, the DPWG guidelines propose different dosing recommendations for 6-TG, according to the presence of genetic polymorphisms in TPMT or NUDT15. Dosing recommendations for thiopurines are summarized in Table I (75).
One of the risks associated with TPMT and NUDT15 genotyping before prescribing thiopurines is the possibility of underdosing, as not all intermediate metabolizers experience myelosuppression at standard doses of thiopurines (82). Therefore, the guidelines also recommend monitoring the efficacy of thiopurine treatment and further adjusting the dose once the steady state is reached and according to routinely monitored clinical parameters such as leukocyte count (11, 75, 77).
Besides TPMT genotype tests, certain clinical laboratories offer also TPMT activity determination. Measuring TPMT activity has the advantage of identifying patients with high TPMT activity, and those who have low TPMT activity on account of other non-Tier 1 genetic polymorphisms. Assays for testing NUDT15 enzyme activity are usually not integrated into clinical practice (77, 83).
Countries like the US, Canada, the UK, The Netherlands, France, Belgium, Spain, Australia, and New Zealand, highly recommend TPMT and NUDT15 genotyping prior to treatment, due to their significant association with treatment outcomes (84–86). Additionally, St. Jude Children's Research Hospital is currently conducting a clinical trial entitled "Total Therapy XVII for Newly Diagnosed Patients with Acute Lymphoblastic Leukemia and Lymphoma", employing precision medicine strategies based on genomic features to improve cure rates and quality of life for children with ALL, highlighting the forever evolving importance of precision medicine (87).
ANALYTICAL METHODS FOR INDIVIDUALIZATION OF THIOPURINE THERAPY
Determination of TPMT and NUDT15 genotypes
Research studies that reported TPMT and NUDT15 genotyping, employ mainly targeted variant analysis by different DNA amplification methods, including polymerase chain reaction (PCR), allele-specific PCR, PCR-single strand conformation polymorphism (PCR-SSCP), denaturing HPLC (DHPLC), TaqMan genotyping, direct (Sanger) sequencing, restriction fragment length polymorphism (RFLP), multiplex amplification refractory mutation (ARMS) and KASP genotyping (88). Some providers of clinical tests offer also larger sets of analyses, simultaneously detecting several different genetic polymorphisms. These panels include methodologies such as microarrays, pyrosequencing, and next-generation sequencing (e.g., massively parallel sequencing). As of now, the National Library of Medicine under the Genetic Testing Registry lists 71 centers, that provide genetic testing for thiopurines, that are located in the United States, Spain, Portugal, Germany, India, Turkey, Canada, Finland, Slovakia, Austria, Netherlands and South Korea (89).
Measurements of TPMT and NUDT15 activity
Numerous methods have been developed for the determination of TPMT activity. The enzymatic activity of TPMT can be monitored by detecting the formation of methylated products or through the consumption of the methyl donor SAM. The first step in TPMT activity assessment is the incubation of hemolysate or lysate of peripheral blood mononuclear cells with a substrate, 6-MP or 6-TG, and a methyl group donor, SAM. Upon incubation, a methylated product is formed and a methyl donor is consumed. The SAM consumed, or methylated products, 6-MMP or 6-MTG, can be detected in various ways. The enzyme activity is usually normalized and reported in terms of either hemoglobin content, packed red cells, erythrocyte count, or protein content.
The first methods for TPMT activity measurement evolved in the late 1970s and 1990s. These were radiochemical methods that measured the conversion of 6-MP to radioactively labeled 6-MMP, using C14- or H3-labelled-SAM (90–92). At the beginning of the 21st century, a rapid immunoassay for TPMT was invented, using an antibody specific for the methylated product, 6-MMP, that was attached to the sample well (Patent, WO 03/076894 A2). Another immunochemical assay was an ELISA-based assay for measuring TPMT activity (93). However, most methods currently used to assess TPMT activity use high-performance liquid chromatography (HPLC) and liquid chromatography coupled with tandem mass spectrometry (LC-MS/MS) (58, 60, 94–97) The former methods exploit the absorbance of 6-MMP or the fluorescence of 6-MTG. LC-MS/MS methods have been developed to determine TPMT activity with high specificity and lower limits of detection as well as shorter analysis time (97). Specificities and sensitivities of TPMT activity tests to detect heterozygous or homozygous individuals range from 90.9–100.0 % and 13.4–100.0 %, resp. (88).
Thiopurine metabolite measurements
The metabolism of 6-MP is complex, involving numerous enzymes (8). This results in many metabolites, among which 6-TGNs and 6-MMP are the most significant. Monitoring the levels of these metabolites to enhance the efficacy of therapy and reduce adverse effects has been proven useful, especially in the treatment of autoimmune disorders (4, 98–100). Measuring the 6-TGNs and 6-MMP metabolites in erythrocytes facilitates dose adjustments for patients receiving the drug (32), as several clinical studies demonstrated a correlation between the levels of 6-TGN and the efficacy of therapy, as well as a correlation between the levels of 6-MMP and certain toxic effects (2, 13, 34–36).
Measurement of intracellular levels of thiopurine nucleotides in erythrocytes is usually assessed by chromatographic methods. Most of the analytical methods measure thiopurine metabolites in erythrocytes (101–103); however, methods using whole blood (104) and a dry blood spot (105) were also developed. The hemolysates from whole blood samples should be prepared as soon as possible, due to limited stability, especially of 6-TGN (105–108). The stability of thiopurine metabolites is enhanced if the blood is spotted on appropriate filter papers and dried out completely (105).
After preparation, samples are separated and thiopurines are detected using HPLC- or UPLC-MS/MS methods (101–105, 109). The HPLC-UV method, compared to the LC-MS/MS method, is less expensive but requires more time for analysis and uses larger quantities of the mobile phase. The advantages of the LC-MS/MS method include shorter time of sample analysis and improved specificity, accuracy, and sensitivity (44).
FUTURE PERSPECTIVES
All three approaches, pharmacogenetics, phenotyping, and therapeutic drug monitoring, are complementary and exhibit great potential to significantly improve patient care by predicting and preventing adverse drug reactions. However, the extent of the implementation of different strategies for individualization of thiopurine dosing varies between different regions and different medical specialties and is difficult to estimate due to the lack of studies. It ranges from 67 % in the UK to 43 % globally (110), from 94 % in dermatology, 50 % in gastroenterology to 20 % in other immune-related conditions (110, 111). Some studies report that the patients are more likely to have their TPMT phenotype determined, compared to the TPMT genotype (112). However, due to the lack of studies, it is hard to define the exact extent of the use of TPMT activity measurements.
A recent systematic review of the cost-effectiveness of pharmacogenomics found that of 11 studies examining the pharmacoeconomic impact of these tests, eight demonstrated cost-effectiveness or even cost-savings associated with pharmacogenomic testing in thiopurine therapy (113). However, to improve the implementation of TPMT and NUDT15 testing prior to treatment initiation, evidently several challenges need to be overcome. These include the need for multidisciplinary teams consisting of physicians, pharmacists, and other healthcare professionals to work together, all of whom need to be trained in the pharmacogenomic intricacies. In addition, the integration of pharmacogenomic data into electronic health records and decision support systems is essential for an efficient workflow but requires significant resources. The issue of reimbursement and the high costs associated with genetic testing are significant barriers, especially in less affluent regions. Finally, standardization is critical to ensure consistent and reliable pharmacogenomic practices.
Table I. CPIC and DPWG recommendations on thiopurine dosing based on TPMT and NUDT15 metabolizer type, for Tier 1 variant alleles (11, 75, 77)
6-MP – 6-mercaptopurine, 6-TG – 6-thioguanine, AZA – azathioprine, CPIC – Clinical pharmacogenetics implementation consortium, DPWG – Dutch Pharmacogenetics Working Group, NUDT15 – nudix hydrolase 15, TPMT – thiopurine S-methyltransferase
Table II. Frequencies of TPMT and NUDT15 variant alleles in biogeographical groups, based on joint consensus of AMP, CPIC, CAP, DPWG, ESPPT, PharmGKB for Tier 1 (TPMT*2, TPMT*3A, TPMT*3B, TPMT*3C, NUDT15*3) and Tier 2 (TPMT*11, TPMT*29, TPMT*42, NUDT15*2, NUDT15*4, NUDT15*6, NUDT15*9, NUDT15*14) (75, 77, 116, 117)
NUDT15 – nudix hydroase 15, TPMT – thiopurine S-methyltransferase; bold – populations with the allele frequency higher than 1 %.
Demand for population-specific guidelines
The need for population-specific pharmacogenomic guidelines for thiopurine treatment is particularly emphasized by the different allele frequencies of genetic polymorphisms in genes such as TPMT and NUDT15 in different populations (Table II). The prevalence of TPMT and NUDT15 polymorphisms affecting thiopurine metabolism and toxicity varies widely among Caucasians, Asians, and Africans (114, 115). According to guidelines, NUDT15 genotyping is more relevant for individuals of East Asian and Hispanic descent with Native American ancestry, whereas TPMT genotyping is important among European and African populations (66).
Additionally, certain populations may be under-researched, so we may lack information on important genetic variants that influence the efficacy of thiopurines and the risk of adverse effects. Consequently, research findings from one population may not be directly applicable to another, underscoring the importance of complementary studies and the development of tailored pharmacogenomic guidelines to optimize therapeutic outcomes, minimize toxicity, and improve the efficacy of thiopurine therapy.
Emerging research in the field of thiopurine pharmacogenomics
The genetic polymorphisms in TPMT and NUDT15 listed in Table I are the only variants clinically annotated with the highest level of evidence for pharmacogenetic testing (11, 77). However, the incomplete concordance between TPMT and NUDT15 genotype or activity and response to thiopurines made it necessary to expand the portfolio of new biomarkers that could be used for individualization of thiopurine therapy (60). It was recognized that other genetic traits, such as genetic polymorphisms in PACSIN2, ITPA, ABCC4, MTHFR, NT5C2, SLC291A, and SLC28A3, influence the efficacy and safety of thiopurines.
PACSIN2 encodes a protein involved in linking the actin cytoskeleton to vesicle formation by regulating tubulin polymerization (118) and regulation of the migration behavior of malignant cells (119). In ALL patients, the homozygous genotype (TT) of the variant rs2413739 (C>T) in the intronic region of PACSIN2 was associated with lower TPMT activity compared to patients without this change (120, 121). The modulation of TPMT activity by PACSIN2 has also been validated in in vitro studies (120). The TT genotype for rs2413739 in PACSIN2 has been associated with thiopurine-related hematotoxicity, hepatotoxicity, and gastrointestinal toxicity, particularly in patients with ALL (120–122); however, the mechanism of interaction between PACSIN2 and TPMT is not yet fully understood.
The nucleotidase NT5C2 dephosphorylates purine monophosphate nucleotides and is important for cellular nucleic acid homeostasis. The NT5C2 variant has been identified as one of the most common genomic lesions specific for relapsed ALL and has been directly linked to thiopurine resistance (123–126). Germline mutations (rs72846714) (124, 125) and acquired somatic mutations (127) were associated with an increased incidence of relapse in ALL patients.
Another candidate considered as a potential pharmacogenetic marker that may influence metabolism and 6-MP-induced toxicities is inosine triphosphate pyrophosphatase (ITPA); however, results correlating its genetic polymorphisms with thiopurine outcome in ALL or IBD patients are inconsistent. The results of a recent meta-analysis examining ITPA polymorphisms and response to thiopurine in ALL patients show that ITPA 94C>A (rs1127354) is associated with 6-MP-induced neutropenia and hepatotoxicity, but not leukopenia (128). Results in IBD study cohorts are even more inconclusive, with many studies, including meta-analyses, finding no association between ITPA genotype and the development of thiopurine toxicity (129–133). Due to the involvement of the enzyme in thiopurine metabolism, certain ITPA variants (rs6139036, rs1127354) have been reported to affect the metabolic profile of thiopurines (134, 135). The differences in the results of these studies may arise from the different settings of the studies, including population characteristics (130, 131, 133, 136, 137) and the co-occurrence of polymorphisms in TPMT and NUDT15 (138). It has been suggested that the ITPA genotype could serve as a genetic marker for improving risk stratification and therapy individualization in patients with ALL (139).
In addition to genetic biomarkers, metabolic factors have been shown to regulate TPMT activity and consequently could influence thiopurine treatment. Besides its methylation function, SAM plays a crucial role in stabilizing enzymes, and one of them is TPMT (140, 141). In experimental assays, the supplementation with SAM stabilizes TPMT posttranslationally and prevents degradation of variant TPMT (49, 141). Consequently, cells, such as MOLT lymphoblasts, cultured under conditions without SAM show significantly lower TPMT activity (142). Our previous findings further show, that TPMT activity measured in hemolysates positively correlates with the concentration of this cofactor (49, 50). Therefore, the measurement of SAM could contribute to the fine-tuning of thiopurine dosing.
Increased toxicity of thiopurines in patients with ALL has also been associated with polymorphisms that reduce the activity of methylenetetrahydrofolate reductase (MTHFR) (71). Considering the significant involvement of this enzyme in the formation and regulation of SAM, this information indicates that folate metabolism is a potential factor influencing the outcome of thiopurine treatment. The involvement of the folate cycle in TPMT activity and subsequently in the response to thiopurines has also been suggested by genetic polymorphisms in the thymidylate synthase (TYMS) gene (143). These polymorphisms may not only affect TPMT activity but have also been significantly associated with an increased risk of developing lymphopenia in patients with inflammatory bowel disease treated with thiopurines (144).
New technological advances in genetic testing
Recent advances in genetic testing technologies will have a significant impact on pharmacogenomics. The transition from targeted variant analysis to comprehensive pharmacogenomic panels has already enabled the simultaneous examination of multiple genetic markers in different genes, supporting precise drug therapy. Genes involved in the thiopurine response, in particular TPMT, NUDT15, and NT5C2, are included in several pharmacogenomic panels as well as panels for hematologic and solid tumor and cancer genomic profiling offered by different providers of genetic testing around the world (89, 145). The introduction of pharmacogenomic panels (passports) can greatly facilitate personalized medicine and adjust drug selection and dosing to optimize therapeutic outcomes and minimize adverse effects (146, 147). However, the clinical implementation of pharmacogenomics is fraught with challenges, including the complexity of interpreting extensive genetic data, the need for robust clinical guidelines to use this information effectively, concerns about the privacy and security of sensitive genetic information, and regulatory and reimbursement considerations (144, 148, 149). Improvements in analytical methods, software development, and artificial intelligence are likely to increase the functionality and consequently the use of such panels to better and more easily predict drug response (150).
Integrated approaches
Ideally, a futuristic projection on the future prospects of thiopurine therapy would involve an integrative approach in pharmacogenomics that comprehensively brings together multiple factors to personalize thiopurine dose adjustment. In addition to genetic variations in TPMT, NUDT15, and other genes mentioned above, these approaches would include environmental factors, patient characteristics such as age and comorbidities, co-therapy, and the metabolomic profile that includes thiopurine metabolite levels and other biochemical markers such as SAM and possibly folate cycle metabolites. Specific emphasis should be given to drug-drug interactions, as complex thiopurine metabolic pathways offer many targets for concomitantly used drugs. Among them, non-steroid anti-inflammatory drugs, furosemide, and sulfasalazine were reported to even inhibit TPMT (151–153). One of the most studied examples of interaction with thiopurines is allopurinol, which enhances thiopurine efficacy and is already applied together with thiopurines to treat refractory examples of IBD and ALL (154–156). The integration of these various data can be facilitated by sophisticated algorithms or predictive models that summarize and analyze the information to recommend optimized dosing regimens. Steps have already been taken toward such predictive models (157–160); however, the cost-effectiveness of such approaches has to be evaluated.
CONCLUSIONS
Pharmacogenomics and therapeutic drug monitoring (TDM) have greatly improved patient care in thiopurine therapy, addressing individual variability in efficacy and safety. Pharmacogenomics guidelines in personalized dosing by identifying TPMT and NUDT15 genetic variants reduce adverse reactions and optimize outcomes of thiopurine therapy. Risk prediction via pharmacogenomics and enzyme activity assessment enables proactive measures to prevent adverse drug reactions, while TDM ensures optimal drug levels, adjusting dosages based on individual factors. Emerging research in thiopurine pharmacogenomics, together with novel technological approaches in genetic testing, forecasts a new era of precision medicine where treatment is increasingly improved by the comprehensive integration of different data points. As the field continues to evolve, it is crucial to include these innovations, translating the knowledge into actionable strategies that refine patient treatment and elevate the standard of care in medical settings where thiopurines play a critical role.
Acronyms, abbreviations, symbols. – 6-MP – 6-mercaptopurine, 6-MMP – 6-methylmercaptopurine, 6-TdGTP – 6-thiodeoxyguanosine triphosphate, 6-TG – 6-thioguanine, 6-TGN – 6-thioguanine nucleotides, ALL – acute lymphoblastic leukemia, ARMS – multiplex amplification refractory mutation, AZA – azathioprine, CPIC – Clinical Pharmacogenetics Implementation Consortium, CPNDS – Canadian Pharmacogenomics Network for Drug Safety, DHPLC – denaturing HPLC, DPWG – Dutch Pharmacogenetics Working Group, GMPS – guanosine 5’-monophosphate synthase, HPLC – high-performance liquid chromatography, HPRT – hypoxanthine phosphoribosyltransferase, IBD – inflammatory bowel disease, IMPDH – inosine 5’-monophosphate dehydrogenase, ITPA – inosine triphosphate pyrophosphatase, LC-MS/MS – liquid chromatography coupled with tandem mass spectrometry, MTHFR – methylenetetrahydrofolate reductase, MTIMP – 6-methylthioinosine monophosphate, NT5C – nucleotidase, NUDT15 – nudix hydrolase 15, PCR – polymerase chain reaction, PCR-SSCP – PCR-single strand conformation polymorphism, RFLP – restriction fragment length polymorphism, SAM – S-adenosyl methionine, SNP – single nucleotide polymorphism, TDM – therapeutic drug monitoring, TGMP – 6-thioguanosine monophosphate, TGTP – 6-thioguanosine triphosphate, TIMP – 6-thioinosine monophosphate, TPMT – thiopurine S-methyltransferase, TYMS – thymidylate synthase, VNTRs – variable number of tandem repeats, XO – xanthine oxidase
Conflicts of interest. – The authors declare no conflict of interest.
Funding. – This work was supported by Slovenian Research and Innovation Agency [P1-0208, I0-0022, I0-E011], and was conducted under the international infrastructure center EATRIS.
Authors contributions. – Conceptualization, D.U. and I.M.R.; writing, review and editing, D.U., F.P., A.Š. and I.M.R. All authors have read and agreed to the published version of the manuscript.
REFERENCES
55 E. Y. Krynetski, N. F. Krynetskaia, Y. Yanishevski and W. E. Evans, Methylation of mercaptopurine, thioguanine, and their nucleotide metabolites by heterologously expressed human thiopurine S-methyltransferase, Mol. Pharmacol. 47(6) (1995) 1141–1147; https://pubmed.ncbi.nlm.nih.gov/7603453/